
根据每一行与集群平均值的距离来可视化R颜色线中的时间序列
根据每一行与集群平均值的距离来可视化R颜色线中的时间序列
提问于 2022-08-25 19:04:23
我对时间序列基因表达数据进行了基于时间表达的聚类。考虑到在时间趋势上有一些变化,我想想象大多数的线是在哪里得到一个更明显的趋势。我想这样做的一种方法是根据线条与集群的平均趋势有多近来给线条着色,但我不知道如何实现它。
我的数据的一个可复制的例子:
dat <- structure(list(gene_name = c("NIPAL3", "NIPAL3", "NIPAL3", "NIPAL3",
"NIPAL3", "SEMA3F", "SEMA3F", "SEMA3F", "SEMA3F", "SEMA3F", "AOC1",
"AOC1", "AOC1", "AOC1", "AOC1", "ZMYND10", "ZMYND10", "ZMYND10",
"ZMYND10", "ZMYND10", "ACSM3", "ACSM3", "ACSM3", "ACSM3", "ACSM3",
"PDK2", "PDK2", "PDK2", "PDK2", "PDK2", "TMEM132A", "TMEM132A",
"TMEM132A", "TMEM132A", "TMEM132A", "ALDH3B1", "ALDH3B1", "ALDH3B1",
"ALDH3B1", "ALDH3B1", "USH1C", "USH1C", "USH1C", "USH1C", "USH1C",
"NOS2", "NOS2", "NOS2", "NOS2", "NOS2"), Sample.name = c("DMSO_1",
"DMSO_2", "DMSO_24", "DMSO_4", "DMSO_8", "DMSO_1", "DMSO_2",
"DMSO_24", "DMSO_4", "DMSO_8", "DMSO_1", "DMSO_2", "DMSO_24",
"DMSO_4", "DMSO_8", "DMSO_1", "DMSO_2", "DMSO_24", "DMSO_4",
"DMSO_8", "DMSO_1", "DMSO_2", "DMSO_24", "DMSO_4", "DMSO_8",
"DMSO_1", "DMSO_2", "DMSO_24", "DMSO_4", "DMSO_8", "DMSO_1",
"DMSO_2", "DMSO_24", "DMSO_4", "DMSO_8", "DMSO_1", "DMSO_2",
"DMSO_24", "DMSO_4", "DMSO_8", "DMSO_1", "DMSO_2", "DMSO_24",
"DMSO_4", "DMSO_8", "DMSO_1", "DMSO_2", "DMSO_24", "DMSO_4",
"DMSO_8"), Treatment = c("DMSO", "DMSO", "DMSO", "DMSO", "DMSO",
"DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO",
"DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO",
"DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO",
"DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO",
"DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO", "DMSO",
"DMSO", "DMSO", "DMSO", "DMSO", "DMSO"), Time = c(1, 2, 5, 3,
4, 1, 2, 5, 3, 4, 1, 2, 5, 3, 4, 1, 2, 5, 3, 4, 1, 2, 5, 3, 4,
1, 2, 5, 3, 4, 1, 2, 5, 3, 4, 1, 2, 5, 3, 4, 1, 2, 5, 3, 4, 1,
2, 5, 3, 4), counts = c(-0.674728025769495, -1.36397354466049,
0.610379522258895, -1.14928773078363, -0.446571694189183, -0.436985550549815,
-0.499130283673904, -0.450641960945776, -0.827458887288972, -0.245426299345142,
-0.305285624771504, -0.640229241821068, 0.167251080770738, -1.09858244762228,
-0.567447650338103, -0.609472980799307, 0.115975465710009, -2.43581298419879,
-0.085029879459607, -0.604272206569522, -0.559054804869209, -0.827020967548349,
0.168793788002722, -0.951210549139716, 0.15852569469711, -0.724991719902859,
-1.04664568307064, -0.828798242883774, -1.15973081519968, -0.574087099835571,
-0.0620293805158624, -0.235285085548273, -0.830591518382229,
-0.469245592463803, -0.0521555714928603, -0.477982049612678,
-0.59701614315213, 0.208614404395684, -1.00028569852069, -0.503026174159395,
-0.631726558325577, -0.60884838805659, -0.0250059525913151, -0.680997114400299,
-0.249988532737743, -0.856318481580249, -0.811356236427306, -0.48845066252092,
-0.928288203615435, -0.508993568295652)), row.names = c(NA, -50L
), class = c("tbl_df", "tbl", "data.frame"))Gene_name列包含了我用于聚类的基因。样品名称包含测量的生物条件和时间。例如,DMSO_1与DMSO样品在1h时的测量相对应。同样的信息也出现在两个单独的列(即处理和时间)上,计数列对应于一个基因的平均、规模化计数,而簇列是每个基因被分配的簇(总共有8个簇)。
我正在绘制我的数据
dat_r%>%
ggplot(aes(Time, counts)) +
geom_line(aes(group = gene_name, alpha = 0.3)) +
geom_smooth(stat = "summary", fun = "mean") 这是一个集群的例子,我正在使用上面的代码和我的完整数据。
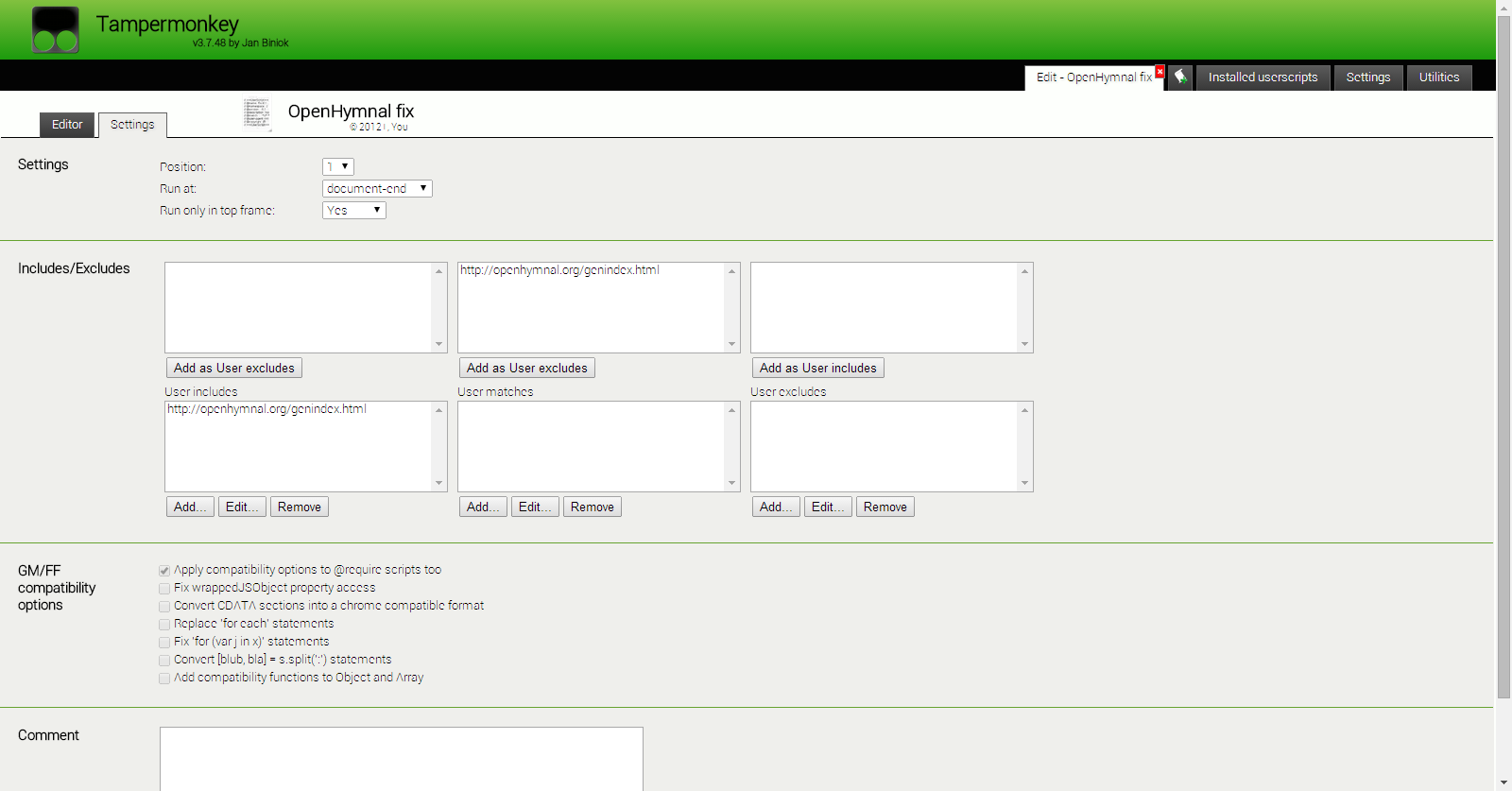
所需输出的一个示例(取自这里)
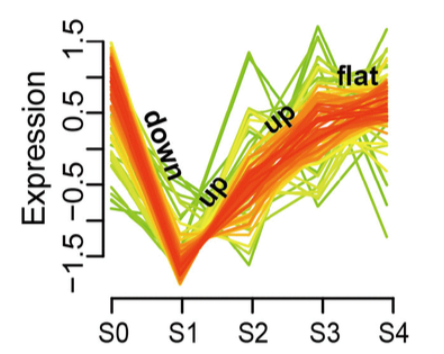
我尝试使用以下代码根据每一行与集群的平均趋势之间的距离对其进行着色。
dat %>%
group_by(gene_name) %>%
mutate(gene_var = counts - mean(counts, na.rm = TRUE)) %>%
ggplot(aes(Time,counts, color = gene_var)) +
geom_line(aes(group = gene_name, alpha = 0.3)) +
scale_color_distiller(palette = "YlOrRd")但是返回的情节并不是用一种颜色来着色线条。
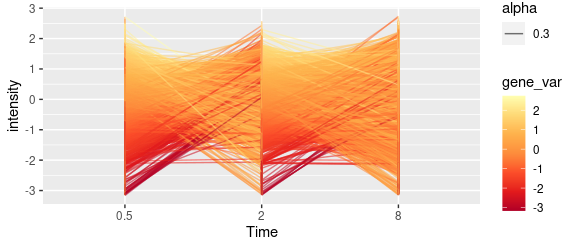
回答 1
Stack Overflow用户
回答已采纳
发布于 2022-08-25 19:20:52
下面是使用来自storms的dplyr数据的一个例子。我计算每一次风暴的RMSE与平均风速的轨迹。也许不是最好的例子,但我认为它能做你想做的事。
# make some example data
storms_sample <- storms %>%
group_by(name, year) %>%
transmute(wind, obs = row_number()) %>%
filter(obs <= 60, max(obs) >= 55)
# calculate RMSE for each storm, and plot based on that
storms_sample %>%
group_by(obs) %>%
mutate(obs_var = wind - mean(wind, na.rm = TRUE)) %>%
group_by(name, year) %>%
mutate(storm_var = sqrt(mean(obs_var^2))) %>%
ungroup() %>% # EDIT - might fix OP's problem
filter(!storm_var %>% between(15,30)) %>% # Edit to highlight extremes
ggplot(aes(obs, wind, group = interaction(name,year), color = storm_var)) +
geom_line(alpha = 0.6) +
scale_color_distiller(palette = "YlOrRd") +
theme_minimal()
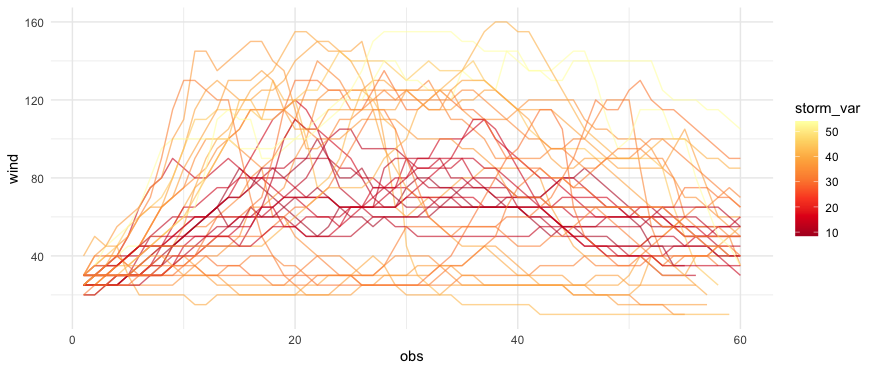
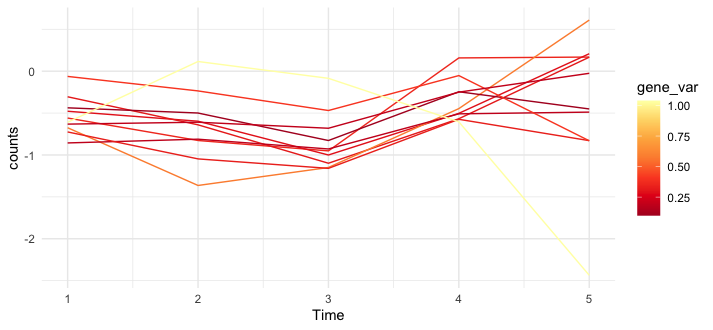
编辑--这是我在新的样本数据上尝试过的代码,它似乎有效--例如,有一个gene_var没有遵循比其他的更黄得多的趋势。
dat %>%
group_by(Time) %>%
mutate(var = counts - mean(counts, na.rm = TRUE)) %>%
group_by(gene_name) %>%
mutate(gene_var = sqrt(mean(var^2))) %>%
ungroup() %>%
ggplot(aes(Time, counts, group = gene_name, color = gene_var)) +
geom_line() +
scale_color_distiller(palette = "YlOrRd") +
theme_minimal()
页面原文内容由Stack Overflow提供。腾讯云小微IT领域专用引擎提供翻译支持
原文链接:
https://stackoverflow.com/questions/73492583
复制相关文章
相似问题
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号