
在R中可视化-一种k-表示聚类发育基因表达数据集
我可以看到很多关于这个话题的帖子,但是没有一个能解决这个问题。如果我错过了一个相关的答案,很抱歉。我有一个大型的蛋白质表达数据集,像这样的样本,如: rep1_0hr,rep1_16hr,rep1_24hr,rep1_48hr,rep1_72hr……
以及行中的2000+蛋白。换句话说,每个样本都是一个不同的开发时间点。
如果感兴趣,则原始数据集是R中的mulvey2015包中的“pRolocdata”,我将其转换为RStudio中的SummarizedExperiment对象。
我首先对数据(一个assay()的SummarizedExperiment数据集)进行k-均值聚类,以获得12个集群:
k_mul <- kmeans(scale(assay(mul)), centers = 12, nstart = 10)然后:
summary(k_mul)产生了预期的产出。
我希望可视化看起来像这样,样本在x轴上,表达式在y轴上。这些图看起来就像使用ggplot中的facet_wrap()生成的:
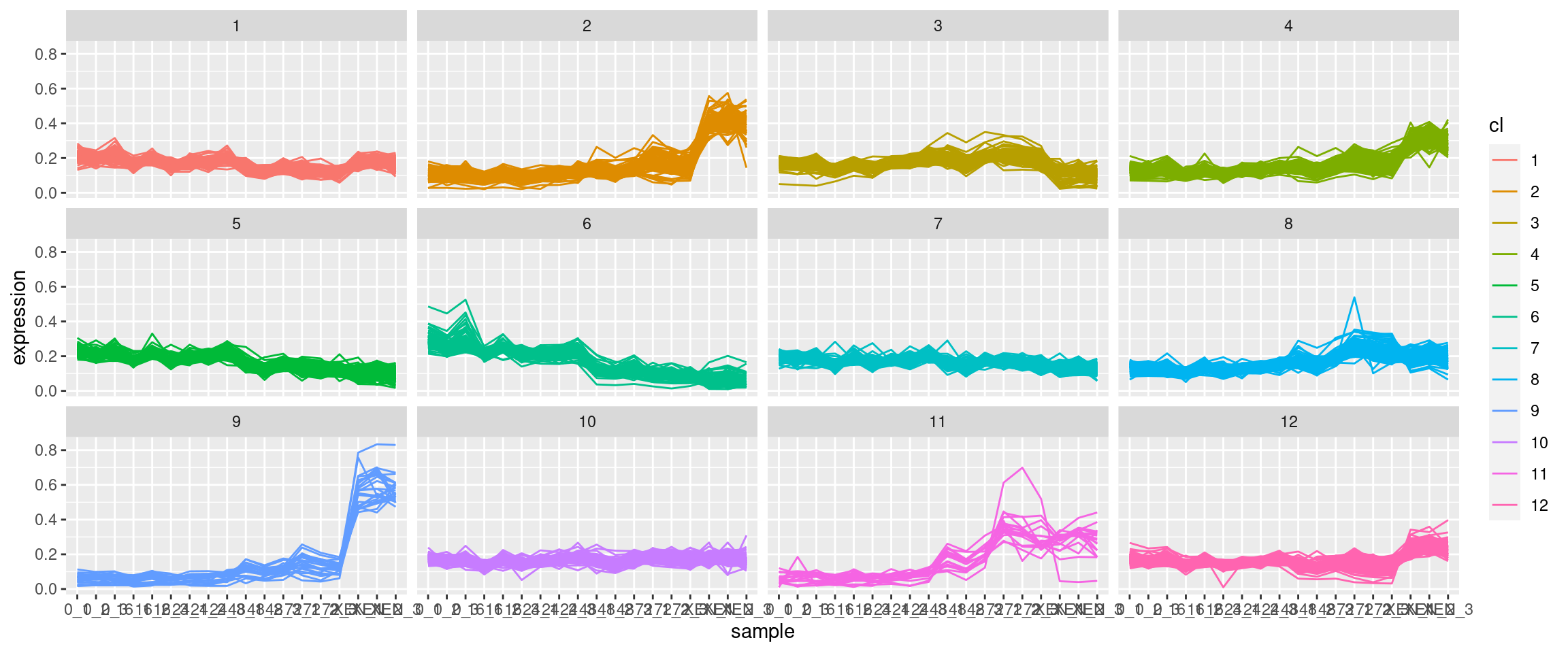
对于ggplot,需要将数据作为带有单个蛋白质集群标识列的数据提供。此外,数据需要采用长格式。我尝试过旋转(pivot_longer)原始数据集,但是当然有大量的数据点。此外,我贴出的图片显示,在任何一幅图中,彩色线条的数量都小于蛋白质的总数,这表明数据集上可能首先出现了维数减少,但我不确定。到目前为止,我一直在运行没有降维的kmeans算法。我能得到如何制作这个情节的指导吗?
回答 2
Stack Overflow用户
发布于 2022-02-25 13:02:56
以下是我试图扭转这一阴谋的企图:
library(pRolocdata)
library(dplyr)
library(tidyverse)
library(magrittr)
library(ggplot2)
mulvey2015 %>%
Biobase::assayData() %>%
magrittr::extract2("exprs") %>%
data.frame(check.names = FALSE) %>%
tibble::rownames_to_column("prot_id") %>%
mutate(.,
cl = kmeans(select(., -prot_id),
centers = 12,
nstart = 10) %>%
magrittr::extract2("cluster") %>%
as.factor()) %>%
pivot_longer(cols = !c(prot_id, cl),
names_to = "Timepoint",
values_to = "Expression") %>%
ggplot(aes(x = Timepoint, y = Expression, color = cl)) +
geom_line(aes(group = prot_id)) +
facet_wrap(~ cl, ncol = 4)至于您的问题,除非pivot_longer无法在键中找到唯一的组合或与数据类型转换相关的问题,否则它通常是很有性能的。该地块可通过以下方式加以改善:
Timepoint
- changing
- 对
geom_lines的alpha参数进行了调整(例如alpha = 0.5),以提供线条密度的概念,为
- 找到一个很好的缩写和顺序,用于
geom_linesaxis.text.x取向
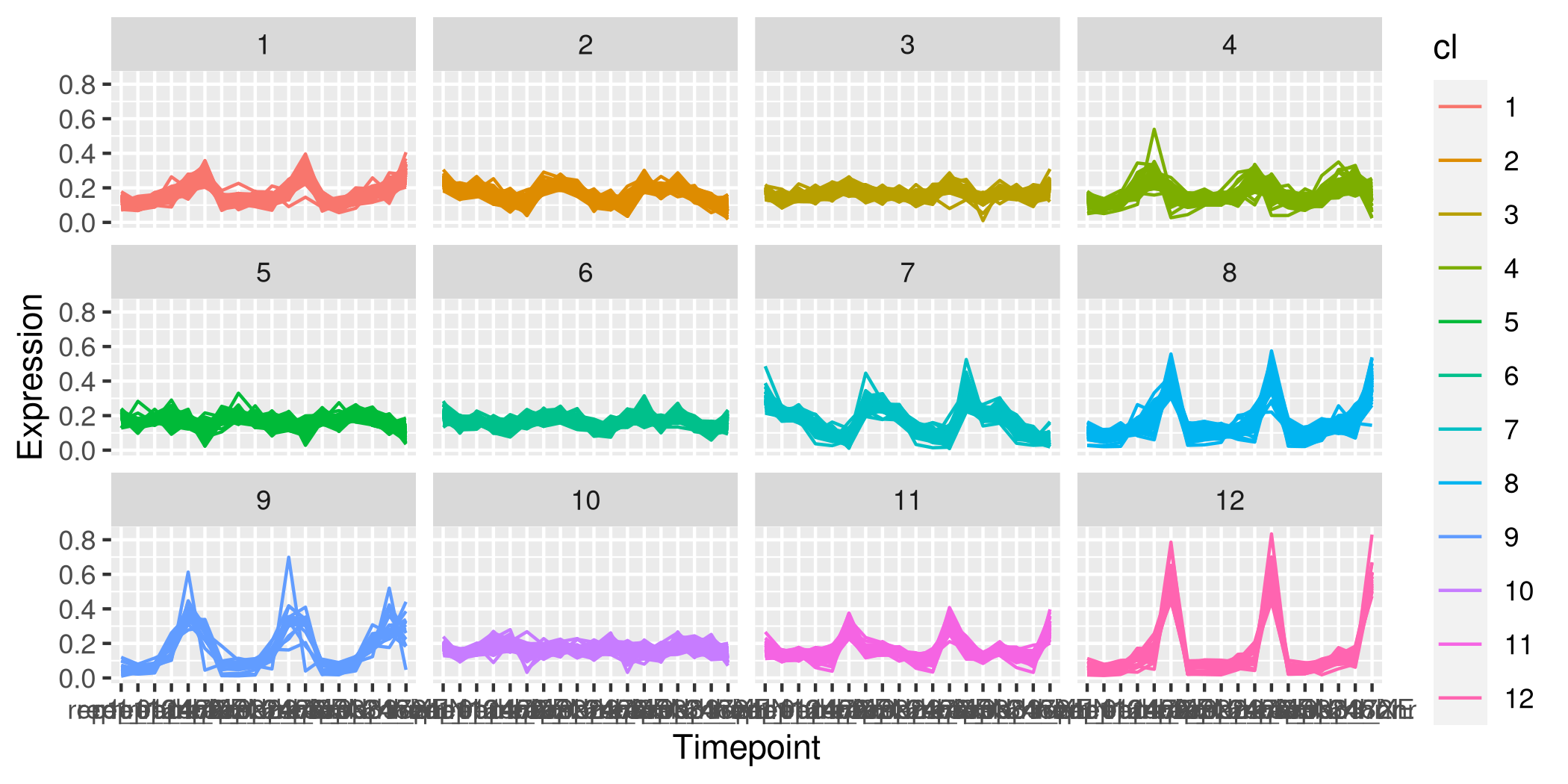
Stack Overflow用户
发布于 2022-02-25 20:57:59
这是我自己的,非常相似的解决方案以上。
dfsa_mul <- data.frame(scale(assay(mul)))
dfsa_mul2 <- rownames_to_column(dfsa_mul, "protID")将kmeans $cluster列添加到dfsa_mul2数据框架中。只在执行clus后将pivot_longer更改为一个因子
dfsa_mul2$clus <- ksa_mul$cluster
dfsa_mul2 %>%
pivot_longer(cols = -c("protID", "clus"),
names_to = "samples",
values_to = "expression") %>%
ggplot(aes(x = samples, y = expression, colour = factor(clus))) +
geom_line(aes(group = protID)) +
facet_wrap(~ factor(clus))这将生成一系列与@sbarbit发布的图表相同的情节。
https://stackoverflow.com/questions/71265540
复制相似问题
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号