数据挖掘—使用seqkit验证SSR位点引物
原创

数据挖掘—使用seqkit验证SSR位点引物
- SSR(简单重复序列) 是基因组中由1–6个碱基组成的串联重复序列,也称为微卫星DNA,具有多态性高、共显性遗传等特点,广泛应用于遗传多样性分析、品种鉴定、分子标记辅助育种等领域
- SSR区域特点是高度重复;高度变异;很少被单独提交到数据库,故文献中很多SSR位点引物在NCBI查不到对应的模版序列,这是正常的
- 在选用文献中的SSR引物时候,不应纠结是否能在数据库检索到,而应该以实验结果为导向
- 如果实在要验证可以下载目标物种的基因组,使用seqkit进行验证,本次以紫苏基因组为例
1.紫苏基因组序列获取
- NCBI中检索紫苏基因组“Perilla frutescens”或“Perilla frutescens (taxid:48386)”中的taxid,优先选取GCF 开头的序列,下载 Genome sequences (FASTA)文件
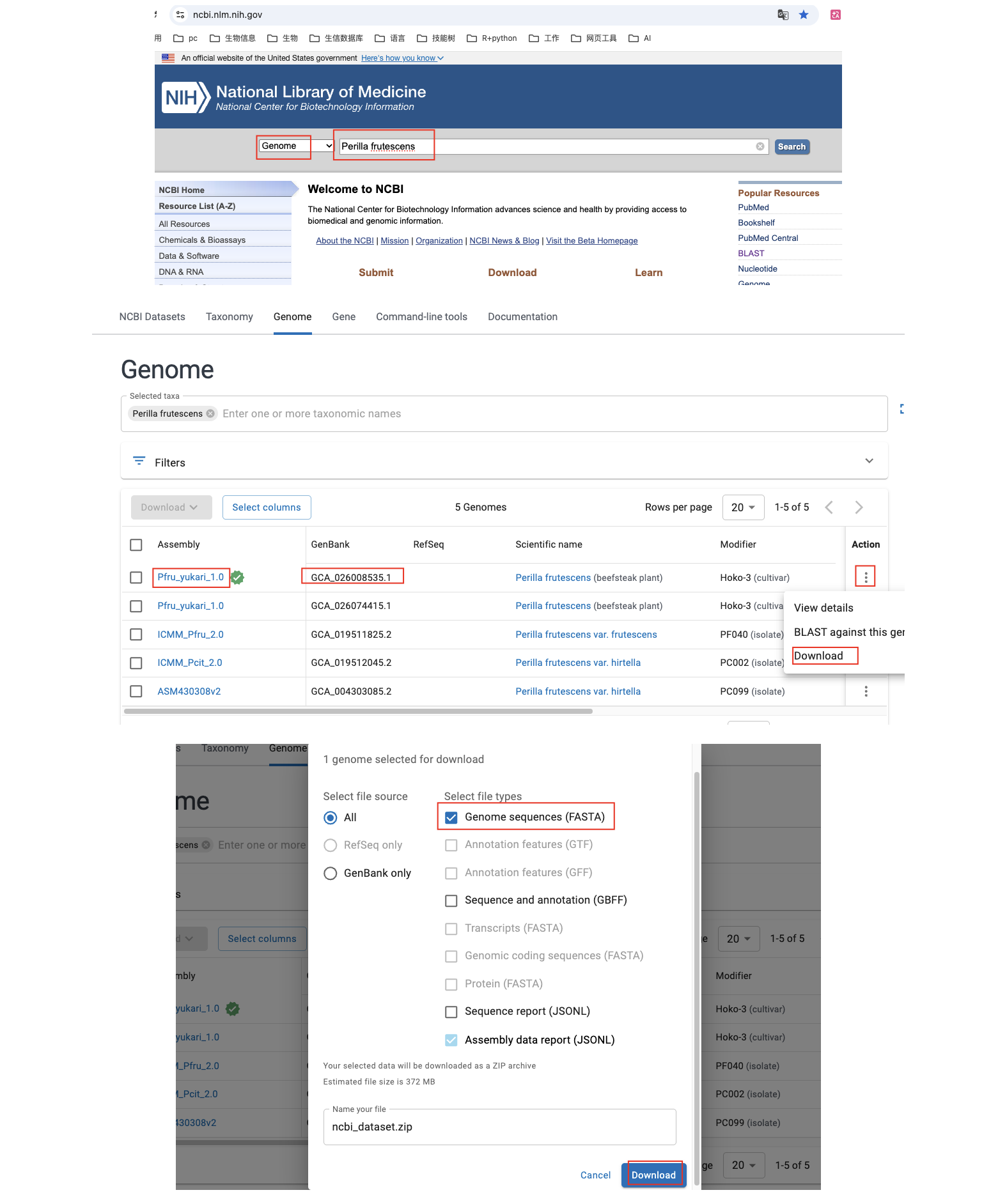
- 基因组的文件还是相对较大的,尽管紫苏的基因组研究较少,没有完全组装好的,也有300来M,解压后,就是我们后续分析需要用到的文件
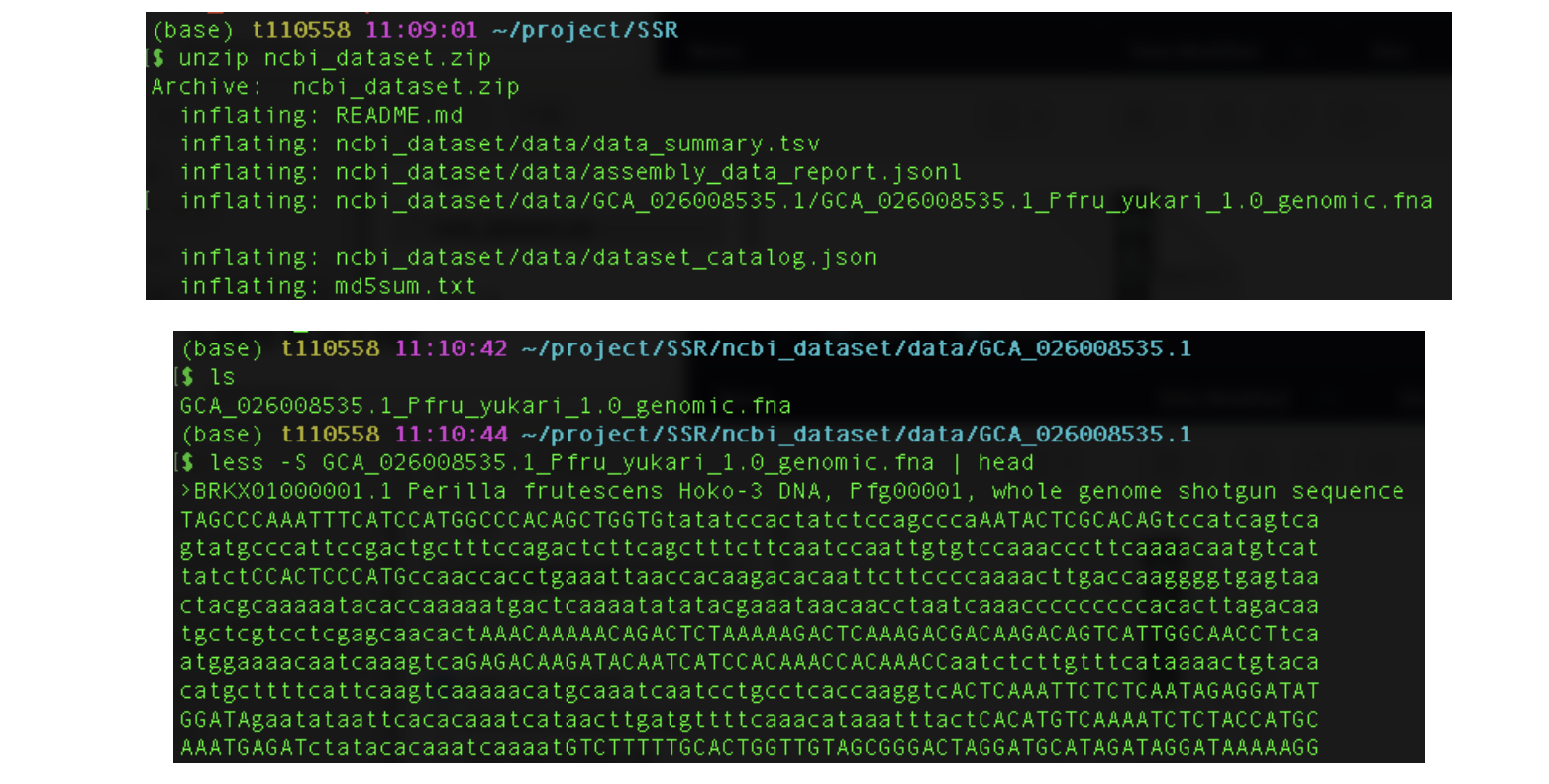
2.SSR位点引物验证
- 引物参考“Identifying SSR markers associated with seed characteristics in Perilla (Perilla frutescens L.)”,
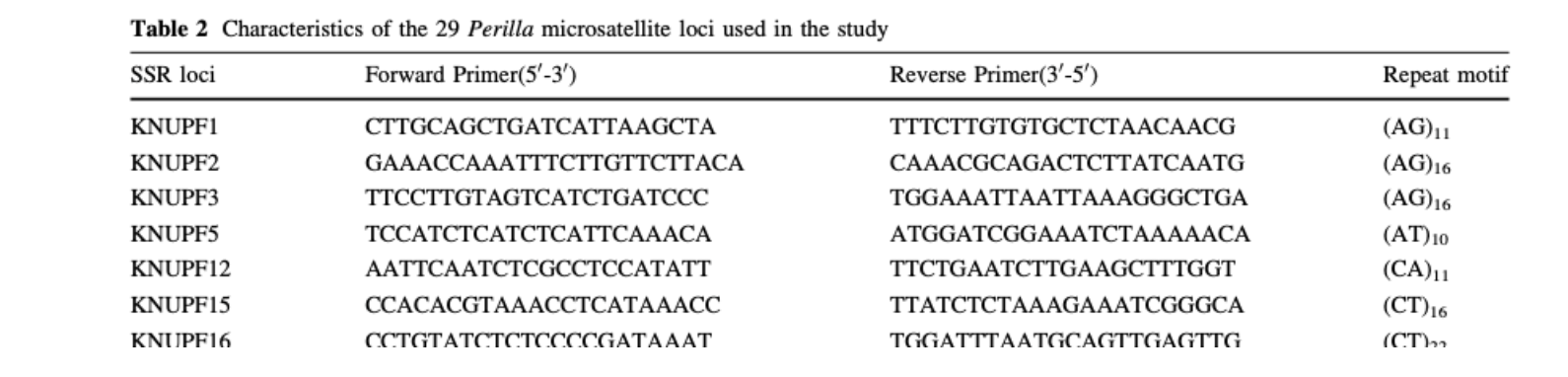
- 以“KNUPF1”位点为例
fw:CTTGCAGCTGATCATTAAGCTA
rv:TTTCTTGTGTGCTCTAACAACG- 使用seqkit进行验证:即这上下游引物都是可以在基因组上检索到的,上游引物在正义链上,下游引物在反义链上。其中下游引物,虽然文献上标注的是(3‘-5'),但是检验出的应该还是(5'-3')的方向,即文献中的上下游引物可以直接使用,下游引物无需在反向互补处理
#conda install seqkit
seqkit locate -i -p CTTGCAGCTGATCATTAAGCTA GCA_026008535.1_Pfru_yukari_1.0_genomic.fna
seqkit locate -i -p TTTCTTGTGTGCTCTAACAACG GCA_026008535.1_Pfru_yukari_1.0_genomic.fna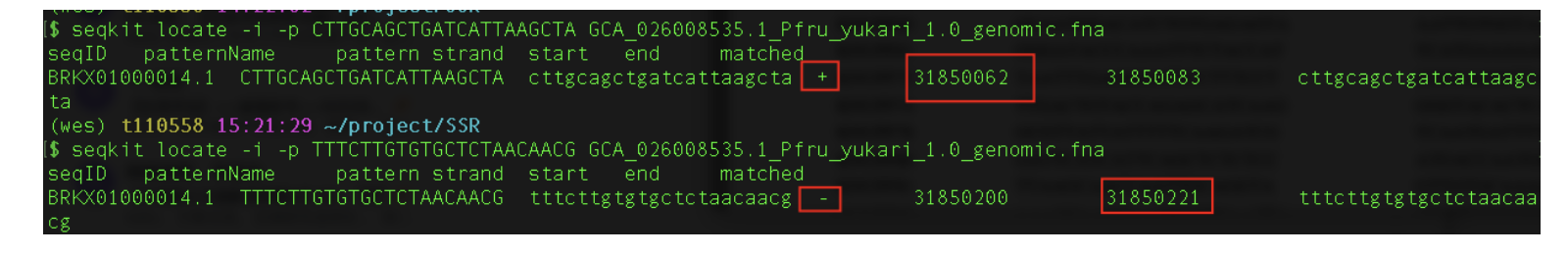
- 提取模版序列
seqkit subseq \
--chr BRKX01000014.1 \
-r 31850062:31850221 \
GCA_026008535.1_Pfru_yukari_1.0_genomic.fna \
-w 0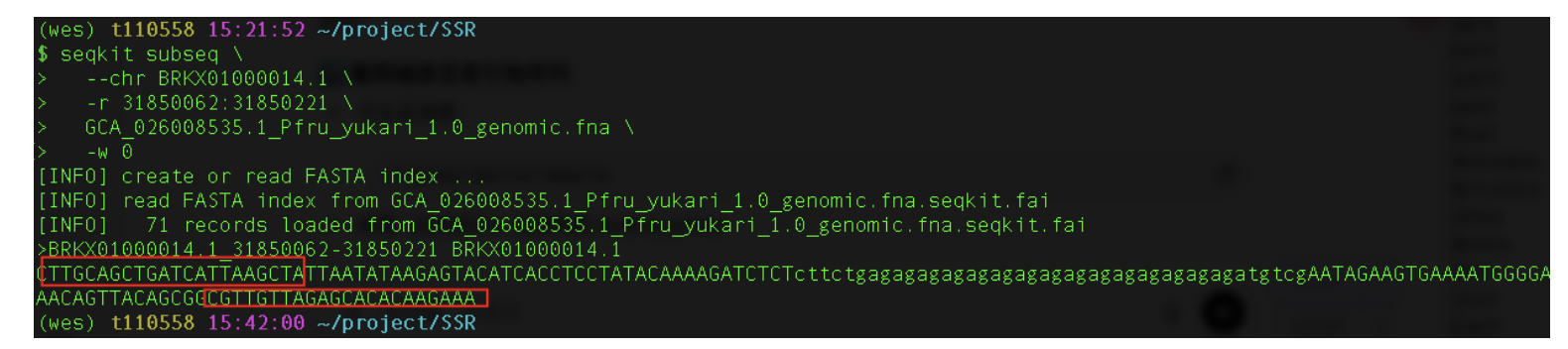
- 查了下中间的序列,≈ (AG)11–12,与文献是高度一致的
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号