单细胞测序—Copykat抽样以及结果映射
原创

单细胞测序—Copykat抽样以及结果映射
- copykat的分析,还是太耗费内存和时间了,详细见单细胞测序—Copykat以及服务器Rstudio运行长脚本的解决方案,因此常规的做法是先抽样,再把结果映射回全部的细胞,这里记录下完整的脚本
rm(list=ls())
options(stringsAsFactors = F)
library(Seurat)
library(ggplot2)
library(clustree)
library(cowplot)
library(data.table)
library(dplyr)
library(stringr)
library(harmony)
#devtools::install_github("navinlabcode/copykat")
library(copykat)
# sce.select <- readRDS('../result/rds/step1_sce_selected_after_umap.rds')
# phe <- sce.select@meta.data
# colnames(phe)
# table(phe$celltype)
mycolors <-c('#E64A35','#4DBBD4' ,'#01A187' ,'#6BD66B','#3C5588' ,'#F29F80' ,
'#8491B6','#91D0C1','#7F5F48','#AF9E85','#4F4FFF','#CE3D33',
'#739B57','#EFE685','#446983','#BB6239','#5DB1DC','#7F2268','#800202','#D8D8CD'
)
############ 提取上皮细胞 ############
# set.seed(12345)
# sce.select.Epithelial <- sce.select[, sce.select$celltype %in% c( 'Epithelial cells' )]
# dim(sce.select.Epithelial)#30558 188187
# saveRDS(
# sce.select.Epithelial,
# "../result/rds/step2_sce_select_Epithelial.rds"
# )
# sce.select.Epithelial <- readRDS('../result/rds/step2_sce_select_Epithelial.rds')
# dim(sce.select.Epithelial)#30558 188187
# table(sce.select.Epithelial$sample)
# Adeno Samples NEC Samples
# 152262 35925
############ 全部上皮细胞 ############
# sce.select.Epithelial <- NormalizeData(sce.select.Epithelial)
# sce.select.Epithelial <- FindVariableFeatures(sce.select.Epithelial)
# sce.select.Epithelial <- ScaleData(sce.select.Epithelial)
# sce.select.Epithelial <- RunPCA(sce.select.Epithelial)
# sce.select.Epithelial <- RunUMAP(sce.select.Epithelial, dims = 1:30)
# saveRDS(
# sce.select.Epithelial,
# "../result/rds/step2_sce_select_Epithelial_after_umap.rds"
# )
message("step1:读取数据")
sce.select.Epithelial <- readRDS('../result/rds/step2_sce_select_Epithelial_after_umap.rds')
table(sce.select.Epithelial@meta.data$sample)
# Adeno Samples NEC Samples
# 152262 35925
############ copyKAT ############
#在子集上训练(跑CopyKAT),然后将结果标签映射回全集
#copykat的作者建议每个样本单独跑,避免批次效应影响CNV推断
#这里我采取了折中的方案,把NEU跑一次,Ade跑一次,最后在合并到一起
#这种方法废弃了,实际操作中,在neu样本中,copykat找不到正常细胞,即来自neu的样本基本上都是肿瘤细胞
#重新采取的方法是neu细胞保留,ade样本抽样,然后合在一起跑copykat
# ############ copyKAT ############
# wd <- getwd()
# message(paste("当前工作目录:",wd))
# table(sce.select.Epithelial$sample)
# # Adeno Samples NEC Samples
# # 152262 35925
# set.seed(123)
# # Adeno抽样
# sce.adeno <- subset(sce.select.Epithelial, subset = sample == "Adeno Samples")
# cells.adeno <- sample(colnames(sce.adeno), 30000)
# # NEC全部保留
# sce.nec <- subset(sce.select.Epithelial, subset = sample == "NEC Samples")
# # 合并
# sce.mix <- subset(
# sce.select.Epithelial,
# cells = c(cells.adeno, colnames(sce.nec))
# )
# counts <- sce.mix[["RNA"]]$counts
# dim(counts)#30558 65925
# counts <- counts[rowSums(counts > 0) > 20, ]
# dim(counts)#24605 65925
#
# message("step2:开始执行copykat")
#
# copykat_result <- copykat(
# rawmat = counts,
# id.type = "S",
# ngene.chr = 3,
# win.size = 50,
# KS.cut = 0.1,
# sam.name = "GBC",
# distance = "euclidean",
# norm.cell.names = "",
# output.seg = FALSE,
# plot.genes = TRUE,
# genome = "hg20",
# n.cores = 12
# )
#
# saveRDS(copykat_result, "../result/rds/step2_sce_select_Epithelial_copykat.rds")
# ############ copyKAT 结果整理 ############
# list.files(pattern = "^GBC")
# #mv GBC* ../result/step2/copykat_result/
# list.files(path= "../result/step2/copykat_result/",pattern = "^GBC")
# [1] "GBC_copykat_clustering_results.rds" "GBC_copykat_CNA_raw_results_gene_by_cell.txt"
# [3] "GBC_copykat_CNA_results.txt" "GBC_copykat_heatmap.jpeg"
# [5] "GBC_copykat_prediction.txt" "GBC_copykat_with_genes_heatmap.pdf"
copykat_result <- readRDS('../result/rds/step2_sce_select_Epithelial_copykat.rds')
pred <- copykat_result$prediction
table(pred$copykat.pred)
# aneuploid diploid not.defined
# 11234 50808 3883
############ copyKAT 结果注释到单细胞中 ############
pred <- copykat_result$prediction
head(pred)
# 只保留明确标签
pred <- pred[pred$copykat.pred %in% c("aneuploid", "diploid"), ]
table(pred$copykat.pred)
# aneuploid diploid
# 11234 50808
#######构建reference Seurat对象######
head(rownames(pred))
cells.use <- rownames(pred)
sce.ref <- subset(
sce.select.Epithelial,
cells = cells.use
)
# 添加标签
sce.ref$copykat <- pred[colnames(sce.ref), "copykat.pred"]
table(sce.ref$copykat)
# aneuploid diploid
# 11234 50808
##这里数量不平衡,导致后续分析多数细胞都被注释为diploid,因此后续需要额外采取抽样处理
set.seed(123)
cells.aneu <- colnames(sce.ref)[sce.ref$copykat == "aneuploid"]
cells.dip <- colnames(sce.ref)[sce.ref$copykat == "diploid"]
cells.dip.sub <- sample(cells.dip, length(cells.aneu))
sce.ref.balanced <- subset(
sce.ref,
cells = c(cells.aneu, cells.dip.sub)
)
table(sce.ref.balanced$copykat)
# aneuploid diploid
# 11234 11234
#######构建构建query(剩余细胞)对象######
cells.all <- colnames(sce.select.Epithelial)
cells.query <- setdiff(cells.all, cells.use)
sce.query <- subset(
sce.select.Epithelial,
cells = cells.query
)
dim(sce.query)
#[1] 30558 126145
#######分开标准化(reference + query象######
# reference
sce.ref.balanced <- NormalizeData(sce.ref.balanced)
sce.ref.balanced <- FindVariableFeatures(sce.ref.balanced, nfeatures = 3000)
sce.ref.balanced <- ScaleData(sce.ref.balanced)
sce.ref.balanced <- RunPCA(sce.ref.balanced, npcs = 30)
# query
sce.query <- NormalizeData(sce.query)
sce.query <- FindVariableFeatures(sce.query, nfeatures = 3000)
sce.query <- ScaleData(sce.query)
sce.query <- RunPCA(sce.query, npcs = 30)
#######Find anchors######
anchors <- FindTransferAnchors(
reference = sce.ref.balanced,
query = sce.query,
dims = 1:30
)
# Performing PCA on the provided reference using 3000 features as input.
# Projecting cell embeddings
# Finding neighborhoods
# Finding anchors
# Found 30064 anchors
#######标签转移######
predictions <- TransferData(
anchorset = anchors,
refdata = sce.ref.balanced$copykat,
dims = 1:30
)
head(predictions)
sce.query$copykat_pred <- predictions$predicted.id
sce.query$copykat_score <- predictions$prediction.score.max
table(sce.query$copykat_pred)
# aneuploid diploid
# 293 125852
#######合并reference + query ######
# reference
sce.ref.balanced$copykat_final <- sce.ref.balanced$copykat
# query
sce.query$copykat_final <- sce.query$copykat_pred
# 合并
sce.all.epi <- merge(sce.ref.balanced, y = sce.query)
table(sce.all.epi$copykat_final)
# aneuploid diploid
# 11527 137086
sce.all.epi
# An object of class Seurat
# 30558 features across 148613 samples within 1 assay
# Active assay: RNA (30558 features, 3000 variable features)
# 6 layers present: counts.1, counts.2, data.1, scale.data.1, data.2, scale.data.2
sce.all.epi <- JoinLayers(sce.all.epi)
sce.all.epi
# An object of class Seurat
# 30558 features across 148613 samples within 1 assay
# Active assay: RNA (30558 features, 3000 variable features)
# 3 layers present: scale.data, data, counts
phe <- sce.all.epi@meta.data
sce.all.epi <- NormalizeData(sce.all.epi)
sce.all.epi <- FindVariableFeatures(sce.all.epi, nfeatures = 3000)
sce.all.epi <- ScaleData(sce.all.epi)
sce.all.epi <- RunPCA(sce.all.epi, npcs = 30)
sce.all.epi <- RunHarmony(sce.all.epi, "sample_name")
sce.all.epi <- RunUMAP(sce.all.epi,reduction = "harmony",dims = 1:30)
saveRDS(sce.all.epi, "../result/rds/step2.1_sce_all_epi_after_copykat_umap.rds")
sce.all.epi <- readRDS('../result/rds/step2.1_sce_all_epi_after_copykat_umap.rds')
table(sce.all.epi$copykat_final)
# aneuploid diploid
# 11527 137086
######可视化 ######
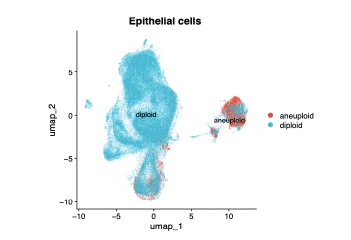
############ umap 可视化 ############
epi_copykat_umap <- DimPlot(
sce.all.epi,
reduction = "umap",
cols = mycolors,
group.by = "copykat_final",
label = TRUE
)+ ggtitle("Epithelial cells ")
epi_copykat_umap
ggsave('../result/step2/epi_copykat_umap.pdf',plot = epi_copykat_umap,width = 6,height = 5)
mycolors1 <- c('#4DBBD4','#E64A35')
epi_sample_umap <- DimPlot(
sce.all.epi,
reduction = "umap",
cols = mycolors1,
group.by = "sample",
label = TRUE
)+ ggtitle("Epithelial cells ")
epi_sample_umap
ggsave('../result/step2/epi_sample_umap.pdf',plot = epi_sample_umap,width = 6,height = 5)
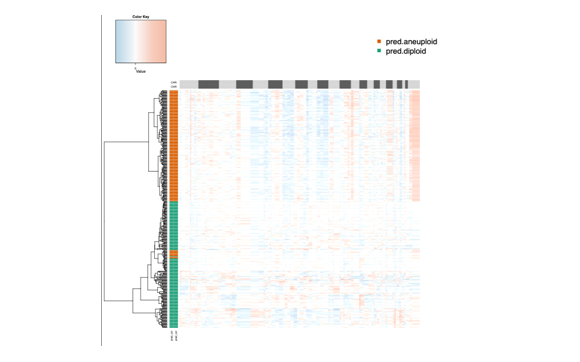
#########copykat 热图绘制#######
CNA.test <- data.frame(copykat_result$CNAmat)
CNA.test[1:4,1:4]
pred.test <- data.frame(copykat_result$prediction)
pred.test <- pred.test[which(pred.test$copykat.pred %in% c("aneuploid","diploid")),] ##keep defined cells
#####细胞数目太多,这里抽样绘制###########
set.seed(123)
# 每类抽样数量
n_sample <- 200
cells_use <- unlist(
lapply(split(rownames(pred.test), pred.test$copykat.pred), function(x){
sample(x, min(length(x), n_sample))
})
)
# 子集数据
CNA.sub <- CNA.test[, c(colnames(CNA.test)[1:3], cells_use)]
pred.sub <- pred.test[cells_use, ]
########## 颜色设置 ##########
# 热图配色
my_palette <- colorRampPalette(rev(RColorBrewer::brewer.pal(n = 3, name = "RdBu")))(n = 999)
# 染色体颜色
chr <- as.numeric(CNA.sub$chrom) %% 2 + 1
rbPal1 <- colorRampPalette(c('black','grey'))
CHR <- rbPal1(2)[as.numeric(chr)]
chr1 <- cbind(CHR, CHR)
# 细胞注释颜色
rbPal5 <- colorRampPalette(RColorBrewer::brewer.pal(n = 8, name = "Dark2")[2:1])
com.preN <- pred.sub$copykat.pred
pred_col <- rbPal5(2)[as.numeric(factor(com.preN))]
cells <- rbind(pred_col, pred_col)
# 颜色断点
col_breaks = c(seq(-1,-0.4,length=50),
seq(-0.4,-0.2,length=150),
seq(-0.2,0.2,length=600),
seq(0.2,0.4,length=150),
seq(0.4,1,length=50))
########## 绘图 ##########
pdf("../result/step2/copykat_result/copykat_balanced_sampling.pdf", height = 15, width = 15)
heatmap.3(
t(CNA.sub[,4:ncol(CNA.sub)]),
dendrogram = "r",
distfun = function(x) parallelDist::parDist(x, threads = 4, method = "euclidean"),
hclustfun = function(x) hclust(x, method = "ward.D2"),
ColSideColors = chr1,
RowSideColors = cells,
Colv = NA,
Rowv = TRUE,
notecol = "black",
col = my_palette,
breaks = col_breaks,
key = TRUE,
keysize = 1,
density.info = "none",
trace = "none",
cexRow = 0.1,
cexCol = 0.1,
cex.main = 1,
cex.lab = 0.1,
symm = FALSE,
symkey = FALSE,
symbreaks = TRUE,
cex = 1,
margins = c(10,10),
labRow = NA,
labCol = NA
)
legend(
"topright",
paste("pred.", names(table(com.preN)), sep=""),
pch = 15,
col = RColorBrewer::brewer.pal(n = 8, name = "Dark2")[2:1],
cex = 2,
bty = "n"
)
dev.off()
#############小提琴图##############
### 转化cnascore数据 ###
# expr <- CNA.test[,4:ncol(CNA.test)]# 前三列是坐标数据
# CNV_score=expr-1# 转换为以0为中心分布的数据
# CNV_score=CNV_score ^ 2# 平方,防止累加的过程中cnv lose和cnv gain相互抵消,造成没有cnv事件发生的假象
# CNV_score=as.data.frame(colMeans(CNV_score))# 以细胞为单位,获得cnv score
# colnames(CNV_score)="CNV_score"# 更改列名
# CNV_score$CB=rownames(CNV_score)# 加入cell barcode列
#
#
# scRNA <- sce.all.epi
# scRNA <- AddMetaData(scRNA,metadata = pred.test)
#
# scRNA$CB <- colnames(scRNA)# 统一cell barcode名称
# CNV_score=CNV_score%>%inner_join(scRNA@meta.data,by="CB")# 合并cnvscore和聚类结果
#
# library(ggplot2)
# library(ggsignif)
# # 按照CNA变异情况分类:
# ggplot(CNV_score,aes(copykat.pred,CNV_score,fill=copykat.pred))+
# geom_violin(alpha=0.4)+ #小提琴图需要一定的透明度
# stat_boxplot(geom="errorbar",position = position_dodge(width = 0.1),width=0.1)+ # 加上误差棒
# geom_boxplot(alpha=0.5,outlier.size=0, size=0.3, width=0.3)+ # 加上箱线图
# scale_fill_manual(values = ggsci::pal_aaas()(3))+ # 填充颜色
# theme_bw()+# 主题为空白
# geom_signif(comparisons = list(c("aneuploid","diploid")
# ),
# step_increase = 0.1,map_signif_level = F)
# 运行日志
step1:读取数据
当前工作目录: /home/data/t080601/Lixiao/TTF/ST/IN2025121601_liuziyu/code
Adeno Samples NEC Samples
152262 35925
[1] 30558 65925
[1] 24605 65925
step2:开始执行copykat
[1] "running copykat v1.1.0"
[1] "step1: read and filter data ..."
[1] "24605 genes, 65925 cells in raw data"
[1] "10051 genes past LOW.DR filtering"
[1] "step 2: annotations gene coordinates ..."
[1] "start annotation ..."
[1] "step 3: smoothing data with dlm ..."
[1] "step 4: measuring baselines ..."
number of iterations= 108
number of iterations= 135
number of iterations= 502
number of iterations= 343
number of iterations= 293
number of iterations= 734
[1] "low confidence in classification"
[1] "cell: 1"
number of iterations= 183
[1] "cell: 2"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 3"
number of iterations= 125
[1] "cell: 4"
number of iterations= 347
[1] "cell: 5"
number of iterations= 318
[1] "cell: 6"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 7"
number of iterations= 107
[1] "cell: 8"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 9"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 10"
number of iterations= 95
[1] "cell: 11"
number of iterations= 350
[1] "cell: 12"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 13"
number of iterations= 166
[1] "cell: 14"
number of iterations= 296
[1] "cell: 15"
number of iterations= 306
[1] "cell: 16"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 17"
number of iterations= 238
[1] "cell: 18"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 19"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 20"
number of iterations= 115
[1] "cell: 21"
number of iterations= 118
[1] "cell: 22"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 23"
number of iterations= 224
[1] "cell: 24"
number of iterations= 121
[1] "cell: 25"
number of iterations= 105
[1] "cell: 26"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 27"
number of iterations= 83
[1] "cell: 28"
number of iterations= 206
[1] "cell: 29"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 30"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 31"
number of iterations= 76
[1] "cell: 32"
number of iterations= 197
[1] "cell: 33"
number of iterations= 244
[1] "cell: 34"
number of iterations= 315
[1] "cell: 35"
number of iterations= 209
[1] "cell: 36"
number of iterations= 149
[1] "cell: 37"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 38"
number of iterations= 313
[1] "cell: 39"
number of iterations= 224
[1] "cell: 40"
number of iterations= 249
[1] "cell: 41"
number of iterations= 120
[1] "cell: 42"
number of iterations= 166
[1] "cell: 43"
number of iterations= 97
[1] "cell: 44"
number of iterations= 371
[1] "cell: 45"
number of iterations= 167
[1] "cell: 46"
number of iterations= 383
[1] "cell: 47"
number of iterations= 165
[1] "cell: 48"
number of iterations= 384
[1] "cell: 49"
number of iterations= 291
[1] "cell: 50"
number of iterations= 205
[1] "cell: 51"
number of iterations= 309
[1] "cell: 52"
number of iterations= 117
[1] "cell: 53"
number of iterations= 408
[1] "cell: 54"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 55"
number of iterations= 214
[1] "cell: 56"
number of iterations= 115
[1] "cell: 57"
number of iterations= 407
[1] "cell: 58"
number of iterations= 108
[1] "cell: 59"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 60"
number of iterations= 135
[1] "cell: 61"
number of iterations= 154
[1] "cell: 62"
number of iterations= 158
[1] "cell: 63"
number of iterations= 164
[1] "cell: 64"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 65"
number of iterations= 131
[1] "cell: 66"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 67"
number of iterations= 151
[1] "cell: 68"
number of iterations= 414
[1] "cell: 69"
number of iterations= 267
[1] "cell: 70"
number of iterations= 498
[1] "cell: 71"
number of iterations= 268
[1] "cell: 72"
number of iterations= 189
[1] "cell: 73"
number of iterations= 137
[1] "cell: 74"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 75"
number of iterations= 234
[1] "cell: 76"
number of iterations= 136
[1] "cell: 77"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 78"
number of iterations= 119
[1] "cell: 79"
number of iterations= 156
[1] "cell: 80"
number of iterations= 147
[1] "cell: 81"
number of iterations= 339
[1] "cell: 82"
number of iterations= 390
[1] "cell: 83"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 84"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 85"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 86"
number of iterations= 379
[1] "cell: 87"
number of iterations= 245
[1] "cell: 88"
number of iterations= 342
[1] "cell: 89"
number of iterations= 394
[1] "cell: 90"
number of iterations= 466
[1] "cell: 91"
number of iterations= 425
[1] "cell: 92"
number of iterations= 324
[1] "cell: 93"
number of iterations= 156
[1] "cell: 94"
number of iterations= 345
[1] "cell: 95"
number of iterations= 304
[1] "cell: 96"
number of iterations= 287
[1] "cell: 97"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 98"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 99"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 100"
number of iterations= 303
[1] "cell: 101"
number of iterations= 350
[1] "cell: 102"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 103"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 104"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 105"
number of iterations= 434
[1] "cell: 106"
number of iterations= 350
[1] "cell: 107"
number of iterations= 124
[1] "cell: 108"
number of iterations= 246
[1] "cell: 109"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 110"
number of iterations= 282
[1] "cell: 111"
number of iterations= 322
[1] "cell: 112"
number of iterations= 143
[1] "cell: 113"
WARNING! NOT CONVERGENT!
number of iterations= 500
[1] "cell: 114"
number of iterations= 102
[1] "cell: 115"
number of iterations= 84
[1] "step 5: segmentation..."
[1] "step 6: convert to genomic bins..."
[1] "step 7: adjust baseline ..."
[1] "step 8: final prediction ..."
[1] "step 9: saving results..."
[1] "step 10: ploting heatmap ..."
Time difference of 2.413455 days
Warning messages:
1: In asMethod(object) :
sparse->dense coercion: allocating vector of size 12.1 GiB
2: In asMethod(object) :
sparse->dense coercion: allocating vector of size 12.1 GiB
3: In asMethod(object) :
sparse->dense coercion: allocating vector of size 4.6 GiB部分结果图
映射回所有的细胞
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号