单细胞测序—Copykat以及服务器Rstudio运行长脚本的解决方案
原创
单细胞测序—Copykat以及服务器Rstudio运行长脚本的解决方案
原创
sheldor没耳朵
修改于 2026-03-23 10:54:12
修改于 2026-03-23 10:54:12
单细胞测序—Copykat以及服务器Rstudio运行长脚本的解决方案
- 最近在做单细胞和空间转录组的联合分析。单细胞分析部分,数据量巨大,约70万的细胞,其中上皮细胞约18万。我需要对上皮细胞进行copykat的分析,这种分析非常耗费计算资源,
- 其中也没有必要对所有的细胞进行copykat,我这里提取了6w细胞进行分析。copykat的作者建议每个样本单独跑copykat后,再把结果合并,以减少批次效应对算法cnv判断的影响。考虑到我实际项目的情况,我是放在一起跑的,这里需要注意下
- 实际分析中,我开始是在服务器网页端的Rstudio进行分析的,这一步耗时太长(>8 h),Rstudio页面长时间不动就会和服务器断开,导致程序运行失败。正确运行耗时长的代码,不应该采取这样的方法,而是封装好脚本,通过ssh,用nohup来运行
如以下的封装好好的step2_epithelial_copykat.R脚本
rm(list=ls())
options(stringsAsFactors = F)
library(Seurat)
library(ggplot2)
library(clustree)
library(cowplot)
library(data.table)
library(dplyr)
library(stringr)
#devtools::install_github("navinlabcode/copykat")
library(copykat)
# sce.select <- readRDS('../result/rds/step1_sce_selected_after_umap.rds')
# phe <- sce.select@meta.data
# colnames(phe)
# table(phe$celltype)
mycolors <-c('#E64A35','#4DBBD4' ,'#01A187' ,'#6BD66B','#3C5588' ,'#F29F80' ,
'#8491B6','#91D0C1','#7F5F48','#AF9E85','#4F4FFF','#CE3D33',
'#739B57','#EFE685','#446983','#BB6239','#5DB1DC','#7F2268','#800202','#D8D8CD'
)
############ 提取上皮细胞 ############
# set.seed(12345)
# sce.select.Epithelial <- sce.select[, sce.select$celltype %in% c( 'Epithelial cells' )]
# dim(sce.select.Epithelial)#30558 188187
# saveRDS(
# sce.select.Epithelial,
# "../result/rds/step2_sce_select_Epithelial.rds"
# )
# sce.select.Epithelial <- readRDS('../result/rds/step2_sce_select_Epithelial.rds')
# dim(sce.select.Epithelial)#30558 188187
# table(sce.select.Epithelial$sample)
# Adeno Samples NEC Samples
# 152262 35925
############ 全部上皮细胞 ############
# sce.select.Epithelial <- NormalizeData(sce.select.Epithelial)
# sce.select.Epithelial <- FindVariableFeatures(sce.select.Epithelial)
# sce.select.Epithelial <- ScaleData(sce.select.Epithelial)
# sce.select.Epithelial <- RunPCA(sce.select.Epithelial)
# sce.select.Epithelial <- RunUMAP(sce.select.Epithelial, dims = 1:30)
# saveRDS(
# sce.select.Epithelial,
# "../result/rds/step2_sce_select_Epithelial_after_umap.rds"
# )
message("step1:读取数据")
sce.select.Epithelial <- readRDS('../result/rds/step2_sce_select_Epithelial_after_umap.rds')
# phe <- sce.select.Epithelial@meta.data
# colnames(phe)
#
# sample_umap <- DimPlot(
# sce.select.Epithelial,
# reduction = "umap",
# cols = mycolors,
# group.by = "sample",
# label = TRUE,
# raster = F #将点转换为栅格图像,便于画图
# )+ ggtitle("Epithelial Cell")
#
# sample_umap
# ggsave('../result/step2/umap_Epithelial_by_samples.pdf',plot = sample_umap,width = 7,height = 6)
############ copyKAT ############
#在子集上训练(跑CopyKAT),然后将结果标签映射回全集
#copykat的作者建议每个样本单独跑,避免批次效应影响CNV推断
#这里我采取了折中的方案,把NEU跑一次,Ade跑一次,最后在合并到一起
#这种方法废弃了,实际操作中,在neu样本中,copykat找不到正常细胞,即来自neu的样本基本上都是肿瘤细胞
#重新采取的方法是neu细胞保留,ade样本抽样,然后合在一起跑copykat
# ############ 以下代码废除 copyKAT NEU############
#
# copykat_out_neu <- "../result/step2/copykat_result/NEU/"
# setwd(copykat_out_neu)
#
# sce.nec <- subset(
# sce.select.Epithelial,
# subset = sample == "NEC Samples"
# )
#
# counts.nec <- sce.nec[["RNA"]]$counts
# dim(counts.nec)#[1] 30558 35925
# counts.nec <- counts.nec[rowSums(counts.nec > 0) > 20, ]
# dim(counts.nec)#22667 35925
#
# copykat_nec <- copykat(
# rawmat = counts.nec,
# id.type = "S",
# ngene.chr = 3,
# win.size = 50,
# KS.cut = 0.1,
# sam.name = "NEC",
# distance = "euclidean",
# norm.cell.names = "",
# output.seg = FALSE,
# plot.genes = TRUE,
# genome = "hg20",
# n.cores = 12
# )
#
# ############ 以下代码废除 copyKAT ADE############
# copykat_out_neu <- "../result/step2/copykat_result/Ade/"
# setwd(copykat_out_neu)
#
# set.seed(123)
#
# sce.adeno <- subset(
# sce.select.Epithelial,
# subset = sample == "Adeno Samples"
# )
#
# # 1 下采样
# cells_use <- sample(colnames(sce.adeno), 35000)
# sce.adeno.sub <- subset(sce.adeno, cells = cells_use)
#
# # 2 提取counts
# counts.adeno <- sce.adeno.sub[["RNA"]]$counts
# dim(counts.adeno)#30558 50000
# # 3 基因过滤
# counts.adeno <- counts.adeno[rowSums(counts.adeno > 0) > 20, ]
# dim(counts.adeno)#23575 50000
# # 4 跑copyKAT
# copykat_result <- copykat(
# rawmat = counts.adeno,
# id.type = "S",
# ngene.chr = 3,
# win.size = 50,
# KS.cut = 0.1,
# sam.name = "GBC",
# distance = "euclidean",
# norm.cell.names = "",
# output.seg = FALSE,
# plot.genes = TRUE,
# genome = "hg20",
# n.cores = 12
# )
# ############ copyKAT ############
wd <- getwd()
message(paste("当前工作目录:",wd))
table(sce.select.Epithelial$sample)
# Adeno Samples NEC Samples
# 152262 35925
set.seed(123)
# Adeno抽样
sce.adeno <- subset(sce.select.Epithelial, subset = sample == "Adeno Samples")
cells.adeno <- sample(colnames(sce.adeno), 30000)
# NEC全部保留
sce.nec <- subset(sce.select.Epithelial, subset = sample == "NEC Samples")
# 合并
sce.mix <- subset(
sce.select.Epithelial,
cells = c(cells.adeno, colnames(sce.nec))
)
counts <- sce.mix[["RNA"]]$counts
dim(counts)#30558 65925
counts <- counts[rowSums(counts > 0) > 20, ]
dim(counts)#24605 65925
message("step2:开始执行copykat")
copykat_result <- copykat(
rawmat = counts,
id.type = "S",
ngene.chr = 3,
win.size = 50,
KS.cut = 0.1,
sam.name = "GBC",
distance = "euclidean",
norm.cell.names = "",
output.seg = FALSE,
plot.genes = TRUE,
genome = "hg20",
n.cores = 12
)
saveRDS(copykat_result, "../result/rds/step2_sce_select_Epithelial_copykat.rds")封装好之后,在工作目录下直接执行
nohup Rscript step2_epithelial_copykat.R > copykat.log 2>&1 &
#查看日志
cat copykat.log
#或
tail -f copykat.log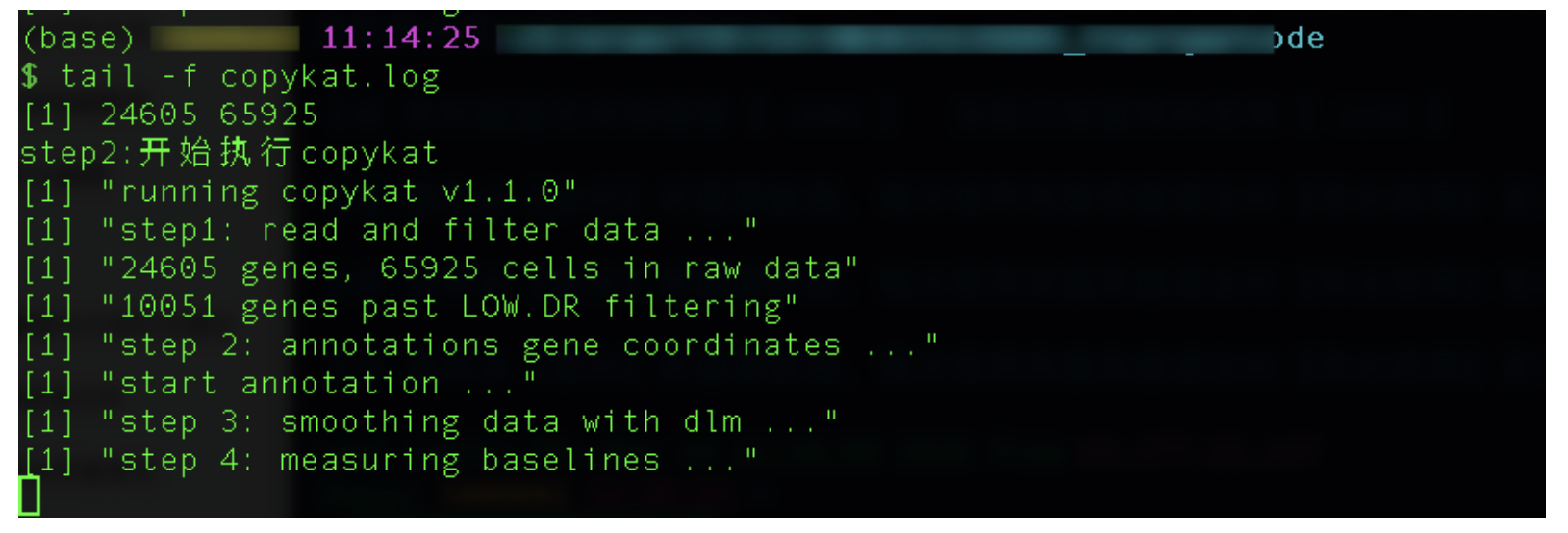
这样放在后台执行,就不会因终端与服务器断开导致程序运行失败了,所以这样才是服务器Rstudio运行长脚本的合理解决方案。以后跑拟时序,cca等耗时长的步骤都需要采取这样的方法
在终端监控该进程
ps fxxw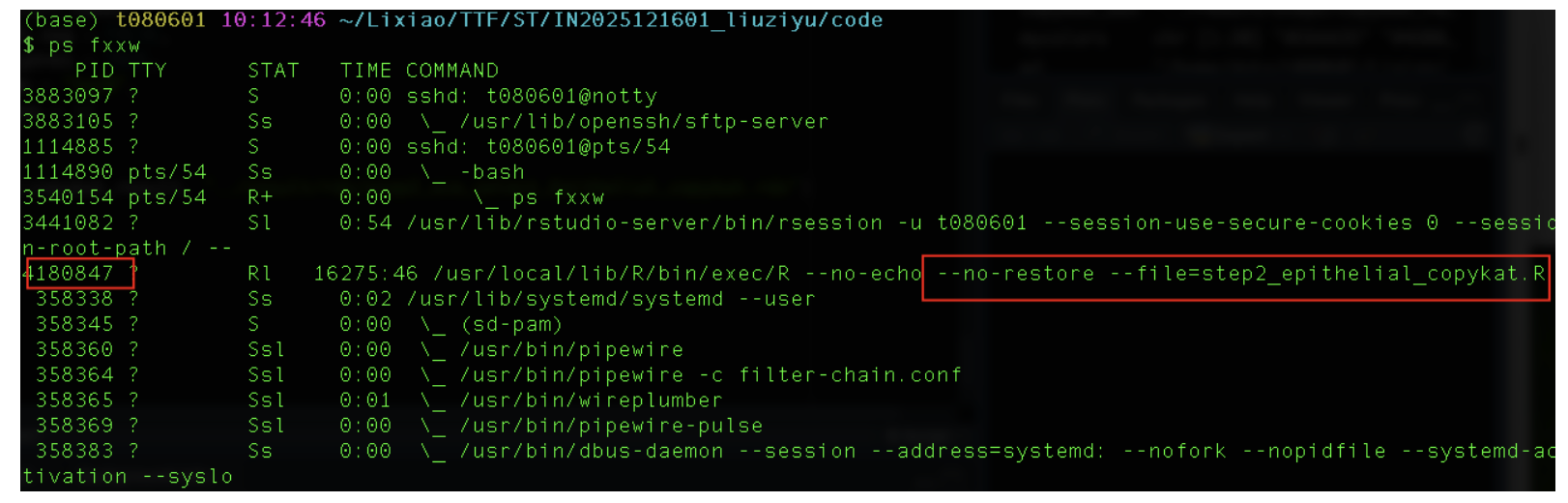
查看该进程已耗时时间,可见即使用10个核心跑,60000个细胞跑了24个小时还是没有跑完,如果不使用这种方式跑,在网页端跑是不可能完成的任务
ps -eo pid,lstart,cmd | grep 4180847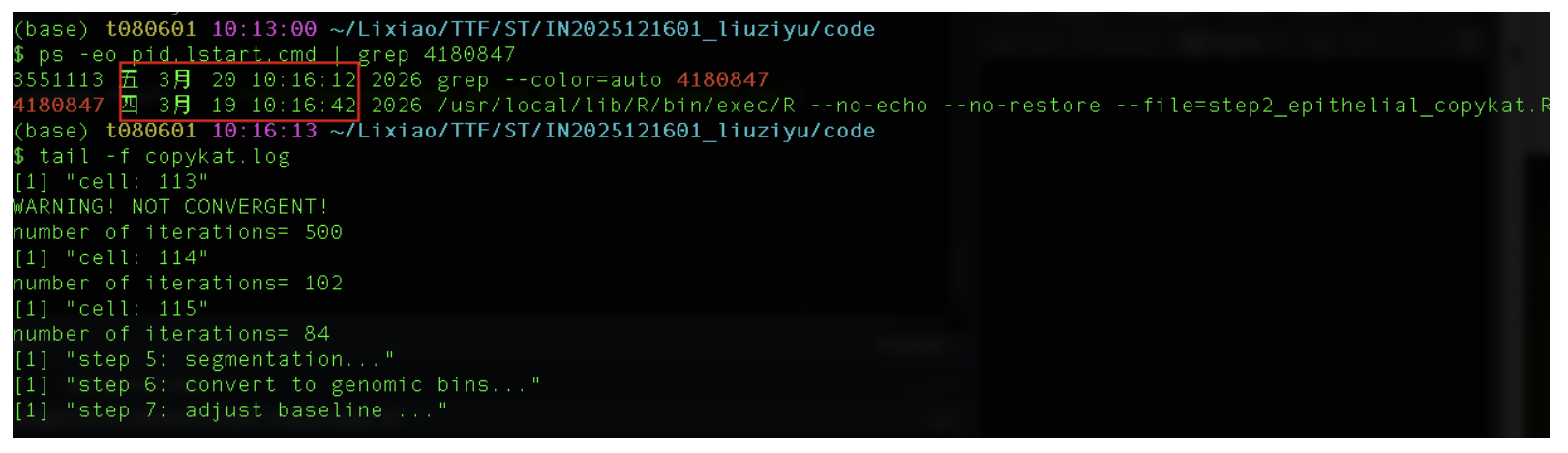
#注:copykat的日志解析
...
number of iterations= 282
[1] "cell: 111"
number of iterations= 322
[1] "cell: 112"
number of iterations= 143
[1] "cell: 113"
WARNING! NOT CONVERGENT!
number of iterations= 500
...
这些日志信息是 CopyKAT 在逐细胞进行贝叶斯分割(Bayesian segmentation)时输出的进度和状态。简单来说,就是算法在逐个细胞地判断:这个细胞的染色体是多了还是少了?
cell: 111,告诉你现在正在处理数据集中的第 111 个细胞
number of iterations= 282: CopyKAT 的核心算法(基于动态线性模型 DLM 的贝叶斯迭代)为了给当前这个细胞找到最合理的拷贝数分段解,迭代了 282 次就达到了收敛标准,找到了一个稳定的解
WARNING! NOT CONVERGENT! + number of iterations= 500:这是一个警告,但不是致命的错误。它表示对于这个特定的细胞(比如第 113 个),算法在跑满了预设的最大迭代次数(这里是 500 次)后,仍然无法找到一个让模型完全稳定的解。最终输出的结果是基于第 500 次迭代的近似值,出现不收敛的情况,
#注2:
...
[1] "cell: 115"
number of iterations= 84
[1] "step 5: segmentation..."
[1] "step 6: convert to genomic bins..."
[1] "step 7: adjust baseline ...,有6万多个细胞,为什么日志才到"cell: 115"
...
CopyKAT 从你的数据中自动选择了一个代表性的细胞子集(可能是基于聚类或随机抽样)来建立基线模型,通常CopyKAT会选取100-200个代表性细胞进行这种详细的贝叶斯迭代,一旦基线模型建立好(完成这115个细胞的迭代后)。在step7中,剩下的6万多个细胞不再需要这种逐细胞的迭代计算,而是基于已经建立的基线模型进行快速推断目前已经跑了29个小时了,有点过久了,占用200G的内存一直再计算,下次再跑,可能需要抽样更少的细胞进行运行,不然实在太耽误时间了。
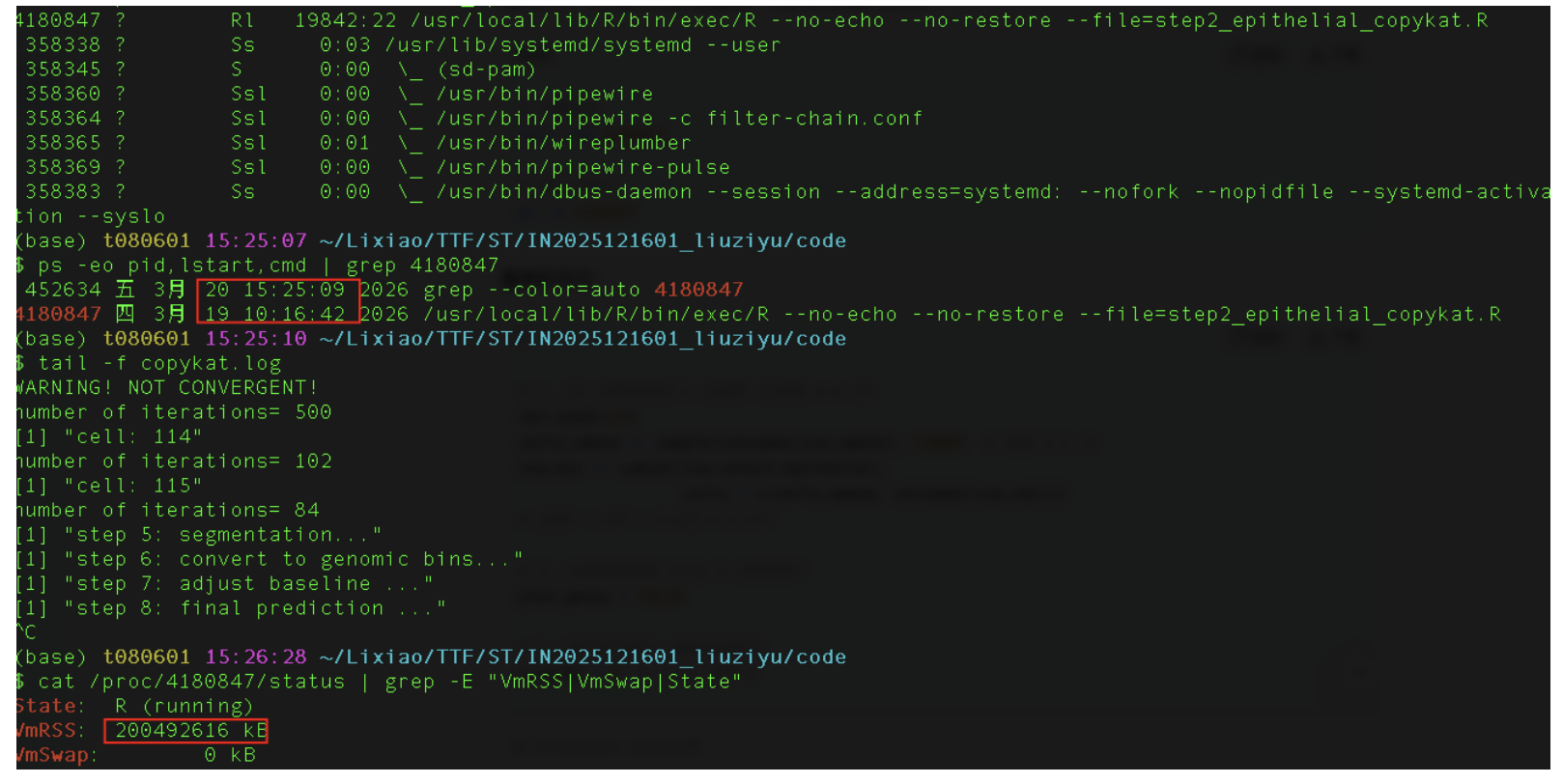
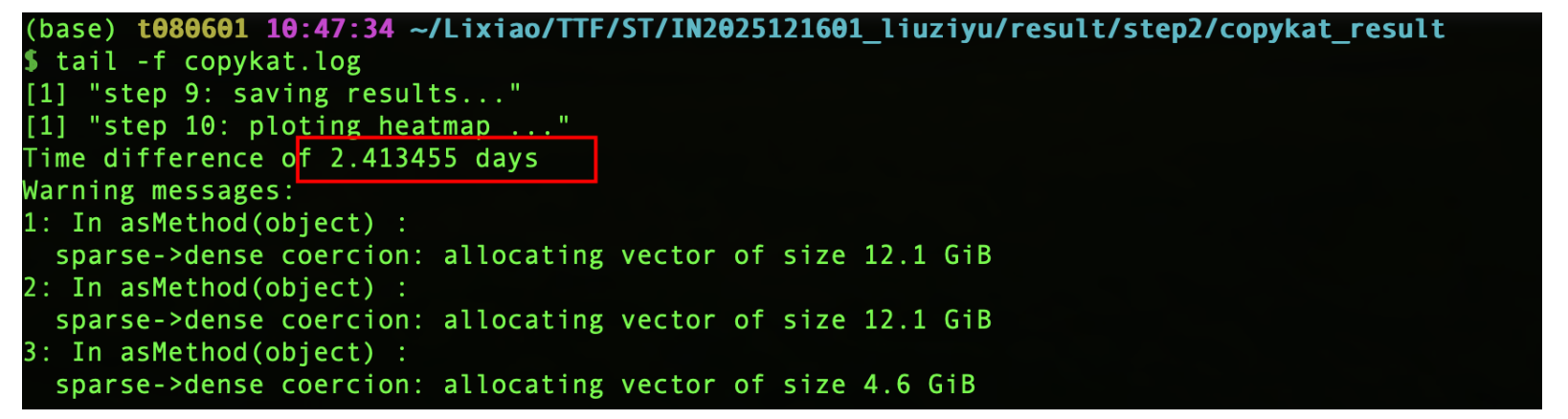
最终跑了2天半
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号