SPATA2【2】之visium HD空转分析及与seurat互联

SPATA2【2】之visium HD空转分析及与seurat互联

KS科研分享与服务-TS的美梦
发布于 2026-03-05 15:55:03
发布于 2026-03-05 15:55:03
偷偷问一下,关注了吗
SPATA2是一款综合性的空间转录组分析工具,能够分析主流的slide、visium以及visium HD数据。我们此前已经介绍过SPATA2对于visium数据的基本分析及与seurat的互联(SPATA2【1】之visium空转分析及与seurat互联),这节介绍一下对于越来越流行的visium HD分析及与Seurat onject的互联。更多关于visium HD数据分析的内容,不论R版还是python版,可以参考往期精彩内容:
空转上游你可以不做,但不能不会呀!详细演示Visium HD上游公共数据库分析及图像对齐矫正
空转联合单细胞分析(五):10X Visium HD上手就是复杂情况?多样本拼片如何进行拆分?
空转联合单细胞分析(六):10X Visium HD seurat V5分析教程
空转联合单细胞分析(八):Visium HD多样本整合分析(基于seurat)
空转联合单细胞分析(九):复现Nature子刊之Visium HD空转RCTD反卷积演示
python版10X空转visium HD分析策略一【scanpy+squidpy】
10X空转visium HD多样本分析策略二【scanpy+spatialdata】
visium HD多样本去批次及sketch下采样分析【scanpy+spatialdata】
1、下载数据及SPATA2读取:
visium HD空转数据自2024年开始,慢慢的越来越流行。相比于visium,其分辨率提高了不止一点。Visium HD 数据来源于以2μm × 2μm 单元标记的空间寡核苷酸。但由于在此分辨率下数据非常稀疏,临近的单元会被合并以生成8μm和16μm的分辨率(spaceranger生成的结果对应有binned_outputs文件夹)。10x 推荐使用8μm合并后的数据进行分析。主流的分析有使用8μm也有16μm的。Visium HD的数据格式与visium是一致的,这里使用的是数据是10X官方的小鼠brain的HD数据,完整的结果下载如下:
# Output Files
wget https://cf.10xgenomics.com/samples/spatial-exp/3.0.0/Visium_HD_Mouse_Brain/Visium_HD_Mouse_Brain_web_summary.html
wget https://cf.10xgenomics.com/samples/spatial-exp/3.0.0/Visium_HD_Mouse_Brain/Visium_HD_Mouse_Brain_cloupe_008um.cloupe
wget https://cf.10xgenomics.com/samples/spatial-exp/3.0.0/Visium_HD_Mouse_Brain/Visium_HD_Mouse_Brain_feature_slice.h5
wget https://cf.10xgenomics.com/samples/spatial-exp/3.0.0/Visium_HD_Mouse_Brain/Visium_HD_Mouse_Brain_metrics_summary.csv
wget https://cf.10xgenomics.com/samples/spatial-exp/3.0.0/Visium_HD_Mouse_Brain/Visium_HD_Mouse_Brain_molecule_info.h5
wget https://cf.10xgenomics.com/samples/spatial-exp/3.0.0/Visium_HD_Mouse_Brain/Visium_HD_Mouse_Brain_spatial.tar.gz
wget https://cf.10xgenomics.com/samples/spatial-exp/3.0.0/Visium_HD_Mouse_Brain/Visium_HD_Mouse_Brain_binned_outputs.tar.gzSPATA2 v3.0也可以进行visium HD数据的读取,使用initiateSpataObjectVisiumHD函数。除了数据读取有些许差别之外,其他方面与visium data的处理没有太多区别。与seurat不同的是,SPATA2读取HD数据,不能像其一样,同时载入多个分辨率的数据,只能选择一个进行分析,这里我们以8um为例。 关于数据的格式:这里的数据是标准的spaceranger output结果,binned_outputs文件夹里面储存了不同分辨率的结果,SPATA2能够根据square_res = ’8um’参数自动找到相对应的文件夹,读取数据。
setwd("~/data_analysis/Spatial_analysis/Visium_HD")
library(SPATA2)
library(tidyverse)object <-
initiateSpataObjectVisiumHD(
sample_name = "MouseBrainH", #Sample name
square_res = '8um',#需要加载的分辨率数据c('16um', '8um', '2um')
mtr = "filtered",#使用filtered的matrix
workers = 1,#线程数
directory_visium = "./hd_data/" #数据所在目录
)## 22:38:03 Initiating SPATA2 object for platform: 'VisiumHD'## 22:38:03 Found ~ binned_outputs\ for resolution 8um.## ## 22:38:12 Active image: 'lowres'.## 22:38:12 Unloading image 'hires'.## 22:38:20 Reading count matrix from '~/binned_outputs/square_008um/filtered_feature_bc_matrix.h5'.## 22:38:56 Active assay: 'gene'.## 22:39:14 Active matrix in assay 'gene': 'counts'## 22:39:14 Identifying tissue outline with `method = obs`.# show overview
object## An object of class SPATA2
## Sample: MouseBrainH
## Size: 393543 x 19059 (spots x molecules)
## Memory: 1.18 Gb
## Platform: VisiumHD (Resolution: 8um)
## Molecular assays (1):
## 1. Assay
## Molecular modality: gene
## Distinct molecules: 19059
## Matrices (1):
## -counts (active)
## Registered images (2):
## - hires (4729x6000 px, not loaded)
## - lowres (473x600 px, active, loaded)
## Meta variables (2): sample, tissue_sectionplotImage(object, img_name = "lowres")
图像处理,识别组织边缘。接下来的步骤与visium的处理别无二致。
# by default, the active image is always used
# you might need to adjust sigma, frgmt_threshold and other parameters to optimize results
object <- identifyPixelContent(object, frgmt_threshold = c(0.01,0.05))object <- identifyTissueOutline(object, method = "image")plotImage(object, outline = TRUE, line_size = 1)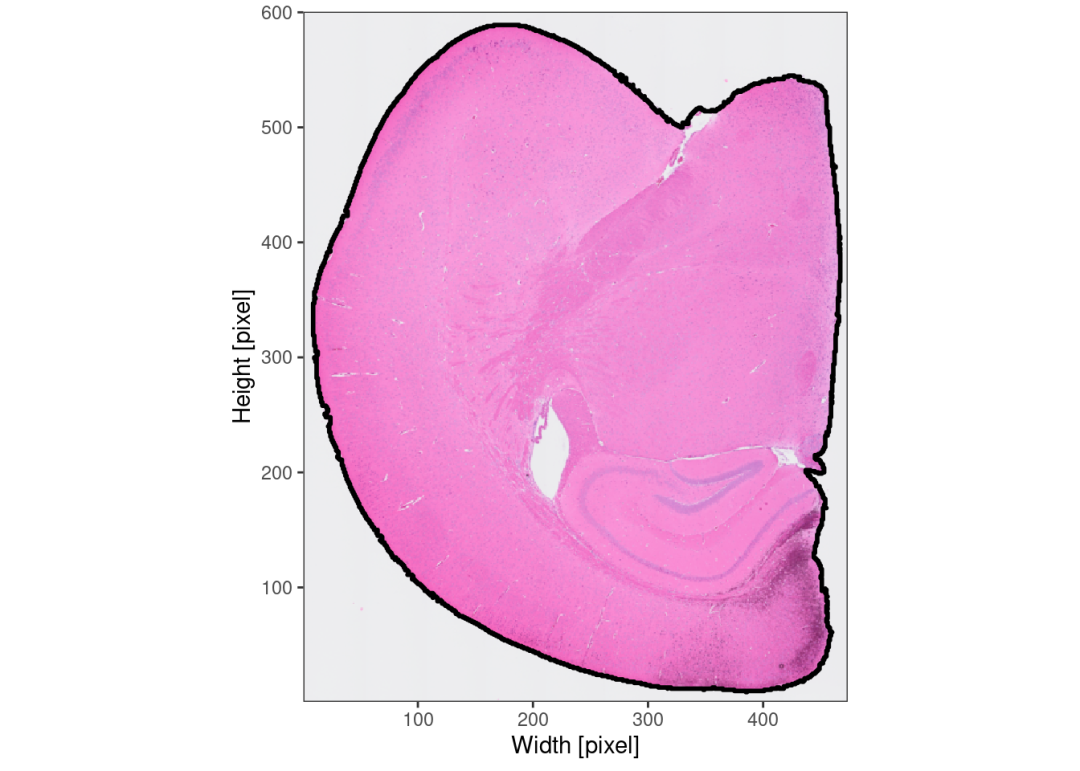
2、seurat读取演示
因为此后的分析包括image、spatial、gene处理都与visium流程是一样的,参考visium的分析,不再重复演示,这里演示一下seurat的读取,然后在后续探究一下visium数据的seurat obj转化为SPATA2 object。以便在seurat处理的数据在SPATA2中进行相关分析。对于seurat读取visium HD数据,需要两个文件夹,一个是spatial,包含图像信息;另外一个是binned_outputs,包含不同分辨率的数据,可以在Load10X_Spatial读取的时候bin.size参数选择需要的分辨率。
library(Seurat)#这里选择16分辨率演示
sce <- Load10X_Spatial(data.dir = './hd_data/', bin.size = c(16))
sce## An object of class Seurat
## 19059 features across 98917 samples within 1 assay
## Active assay: Spatial.016um (19059 features, 0 variable features)
## 1 layer present: counts
## 1 spatial field of view present: slice1.016umSpatialFeaturePlot(sce, features = "nCount_Spatial.016um") + theme(legend.position = "right")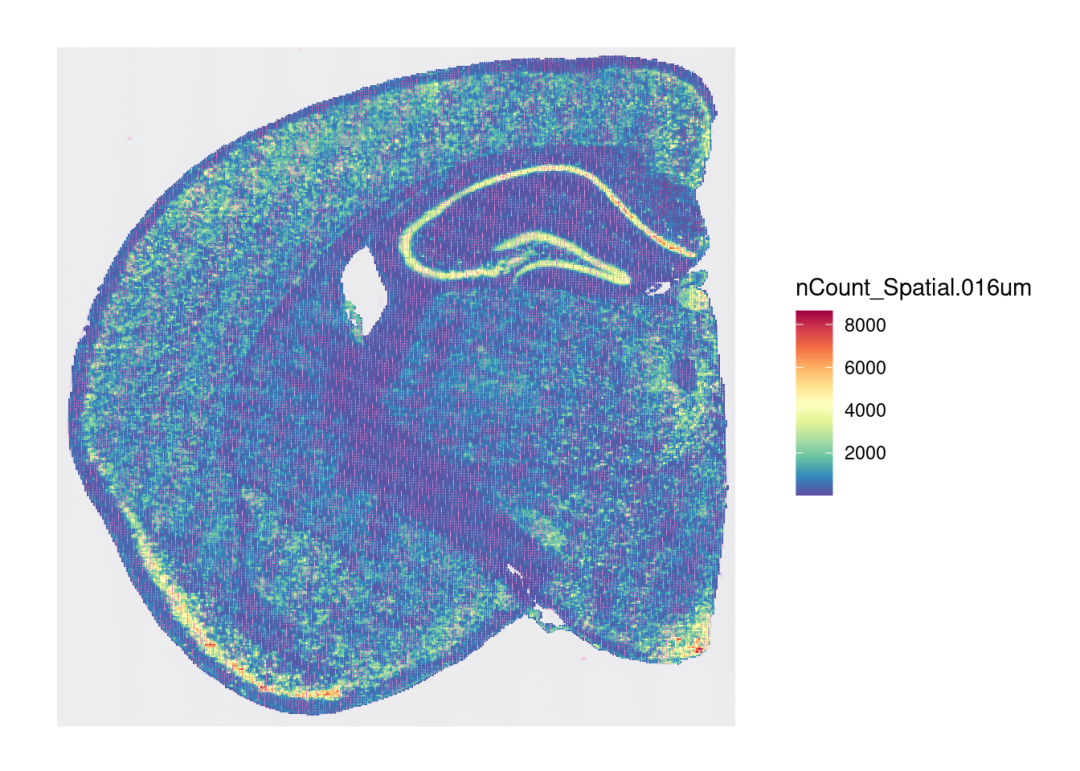
3、visium HD seurat obj转SPATA2 obj
转化也是相当丝滑,没啥问题。
# use assay Spatial
sce_spata2 <-
asSPATA2(
object = sce,
sample_name = "mouse_brain",
platform = "VisiumHD",
img_scale_fct = "lowres",
assay_name = "Spatial.016um",
assay_modality = "gene"
)show(sce_spata2)## An object of class SPATA2
## Sample: mouse_brain
## Size: 98917 x 19059 (spots x molecules)
## Memory: 919.52 Mb
## Platform: VisiumHD (Resolution: 16um)
## Molecular assays (1):
## 1. Assay
## Molecular modality: gene
## Distinct molecules: 19059
## Matrices (1):
## -counts (active)
## Registered images (1):
## - slice1.016um (600x473 px, active, loaded)
## Meta variables (4): sample, orig.ident, nCount_Spatial.016um, nFeature_Spatial.016umplotSurface(sce_spata2, color_by = "nCount_Spatial.016um", pt_size = 1)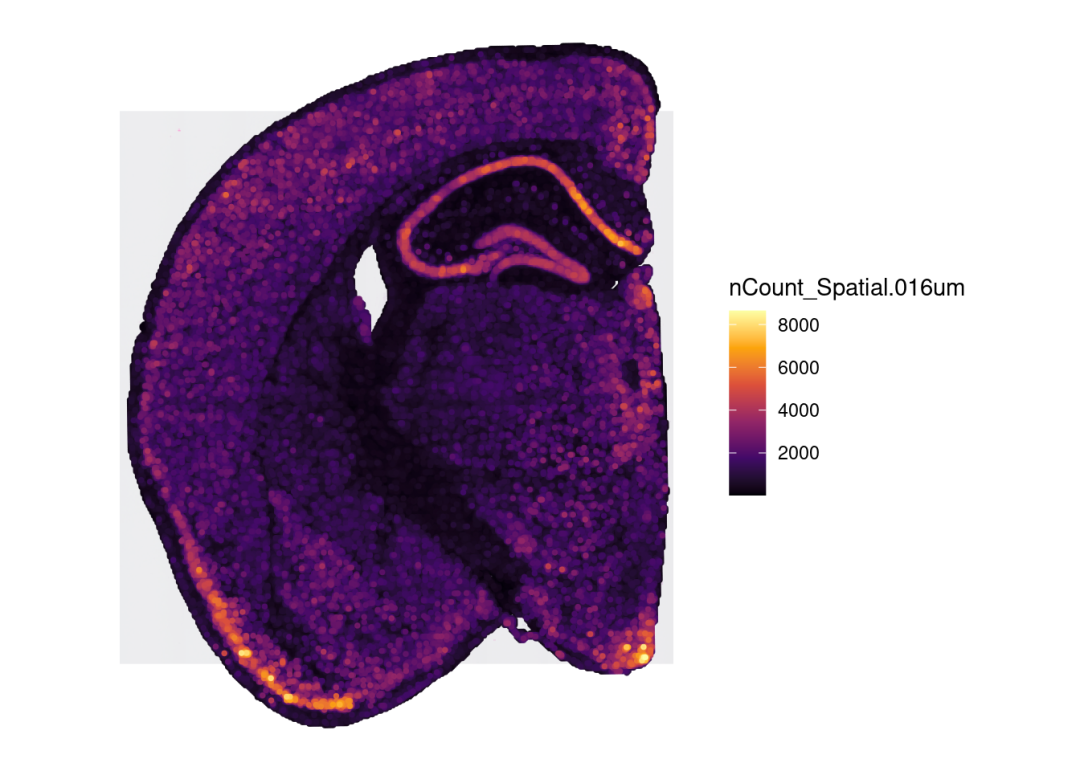
觉得分享有用的点个赞再走呗!
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-03-03,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号