JASPAR分析转录因子与某基因启动子的结合位点及MUT位点
原创
JASPAR分析转录因子与某基因启动子的结合位点及MUT位点
原创
sheldor没耳朵
发布于 2026-01-05 09:49:56
发布于 2026-01-05 09:49:56
JASPAR分析转录因子与某基因启动子的结合位点及MUT位点
最近实验室有个分析需求,要求用JASPAR数据库预测转录因子Sox18与Itch 结合位点(物种:小鼠),需要Itch的启动子区域以及突变后的序列。
如何方便的获取某基因的启动子序列,以及使用JASPAR预测,我已经在之前的帖子中详细记录了
这里主要介绍下,如何找MUT位点,以及后续验证(MUT位点可使用chatgpt辅助,但突变后的序列需通过验证即可)
1.Itch启动子序列获取
- UCSC数据库中检索“Itch”(Mouse),将转录起始位点(TSS)前2000bp序列作为启动子序列(根据基因位于“+”链或“-”链上,向前或向后取2000bp)
- “Itch”位于“+”链
2.结合位点预测
- JASPAR 数据库中获取“Sox18” (MA1563.2) 的核心矩阵(人鼠相同),使用上述启动子 FASTA 序列,设置 Relative profile score threshold = 80% 进行扫描
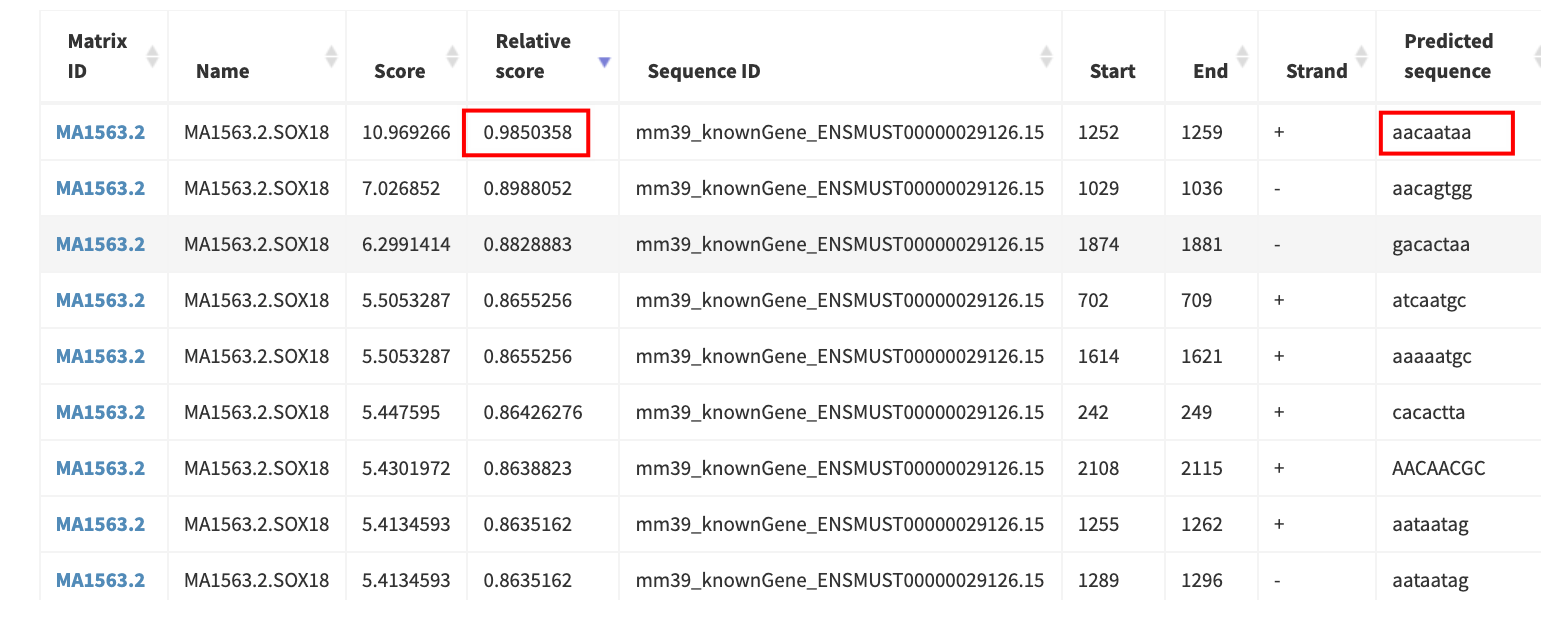
- 综合考虑转录起始位点(TSS)最近的位点和Relative score 较高的位点,选择以下位点
Matrix ID | Name | Score | Relative score | Start | End | Strand | Predicted sequence |
|---|---|---|---|---|---|---|---|
MA1563.2.SOX18 | 10.9693 | 0.98504 | 1252 | 1259 | aacaataa |
- 使用snapgene进行展示
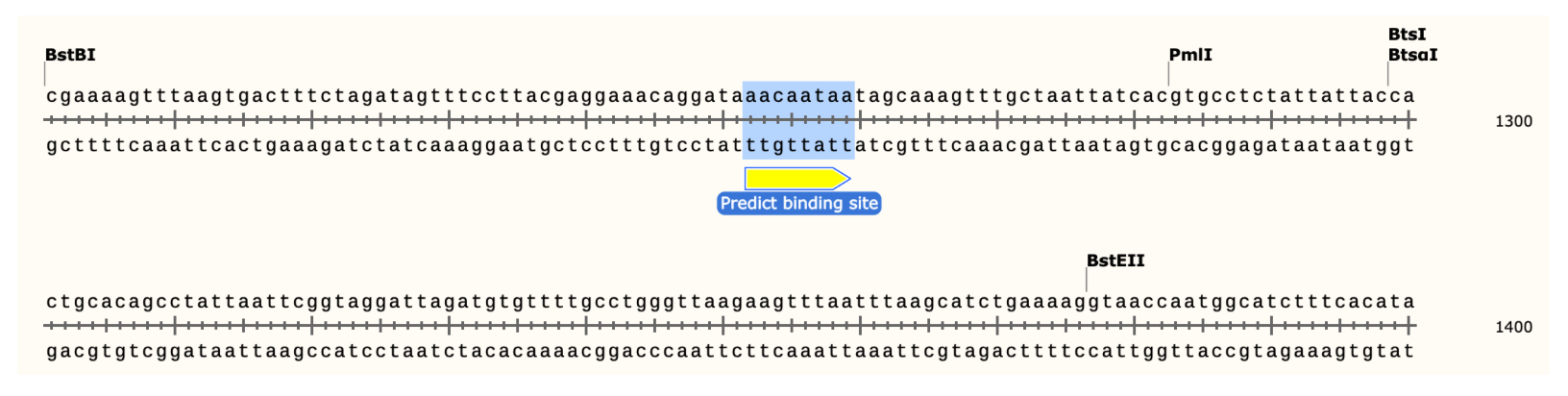
3.Itch-MUT位点
- 2中分析得到其结合位点为WT:5′- AAC AAT AA -3′
- 该位点评分极高,且含有SOX 核心:CAA,距离TSS位点近,结果理想
- MUT位点设计,遵循完全破坏 SOX(HMG-box),不引入新的 TF motif,AT 含量变化合理的原则进行,将“CAA”改为“TTT”MUT: 5′- AAT TTT AA -3′

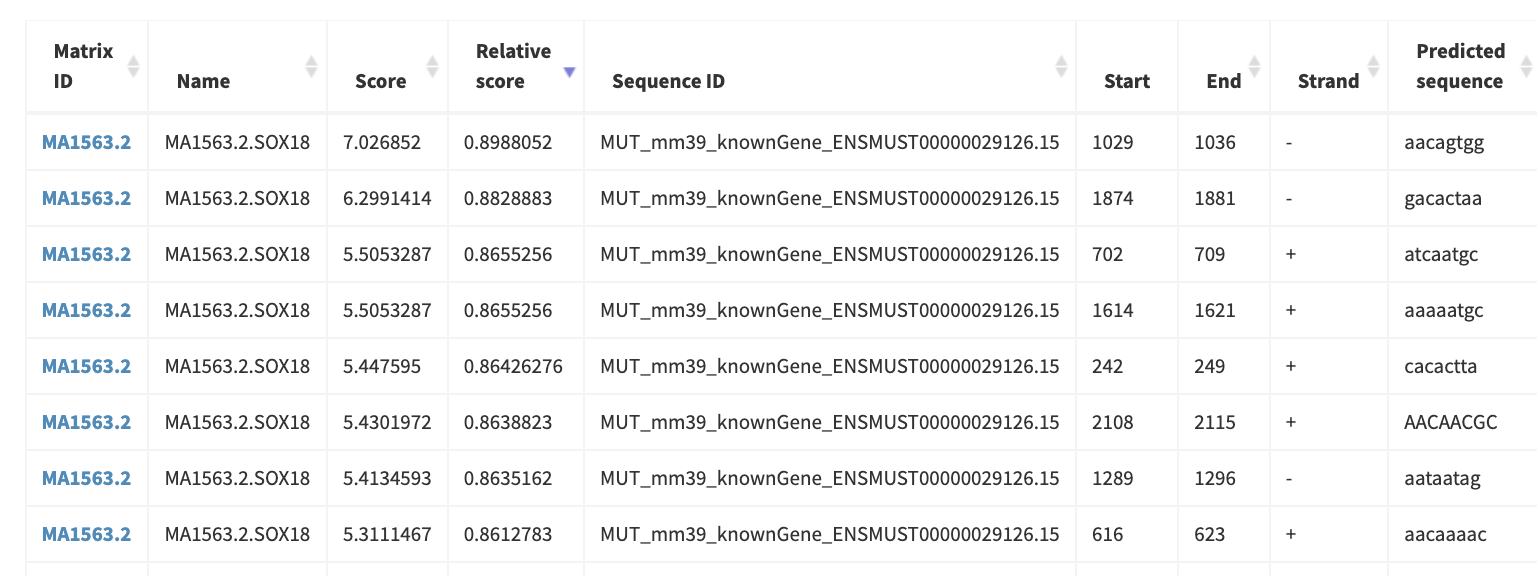
- 验证:将突变后的序列重新使用JASPAR,设置 Relative profile score threshold = 80% 进行扫描,发现1252–1259 这个位点完全消失,且没有接近原位点强度的替代位点,该序列有效
4.相关文件说明
#WT:Itch启动子序列fasta文件,其中小写字母为TSS前2000bp序列,作为启动子区域;大写字母为5‘UTR区域
sup/WT_Itch_promoter_5'UTR.fasta'
#WT:Itch启动子序列,可使用snapgene打开,其中标注了结合位点(可忽略)
sup/WT_Itch_promoter_5'UTR.dna'
#MUT:Itch启动子序列fasta文件,其中小写字母为TSS前2000bp序列,作为启动子区域;大写字母为5‘UTR区域
sup/MUT_Itch_promoter_5'UTR.fasta'
#MUT:Itch启动子序列,可使用snapgene打开,其中标注了结合位点(可忽略)
sup/MUT_Itch_promoter_5'UTR.dna'原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号