单细胞RNA速率-R语言版


今天是生信星球陪你的第1054天
公众号里的文章大多数需要编程基础,如果因为代码看不懂,而跟不上正文的节奏,可以来找我学习,相当于给自己一个新手保护期。我的课程都是循环开课,点进去咨询微信,随时可以报名↓ 生信分析直播课程(每月初开一期) 生信新手保护学习小组长期报名中(每月一期) 单细胞陪伴学习小组(每月一期)
RNA速率(RNA velocity)分析是一种通过比较未剪接(unspliced)和已剪接(spliced)RNA的比例来推断细胞状态转换方向的方法。本教程会介绍如何使用velocyto.R进行RNA速率分析。
其实做RNA速率最好是用python,R版本的实现还是简陋了点儿。且需要注意,velocyto.R只能在服务器和mac上运行,原则上不支持Windows,此处省略n个理由,如果实在不想或者没有服务器可用,可以搜索一下网上的教程,我看到了有办法实现,但还是要花点钱钱,自行搜索一下吧~
1. 环境准备和数据加载
1.1 加载必要的R包
首先清空工作环境,然后加载分析所需的R包。
rm(list = ls())
library(velocyto.R)
library(Seurat)
library(SeuratWrappers)
RhpcBLASctl::blas_set_num_threads(1) # 限制线程数,防止后面的步骤内存溢出
1.2 读取loom文件
loom文件是velocyto输出的标准格式,包含了spliced(已剪接)和unspliced(未剪接)的RNA计数矩阵。这两种RNA的比例可以反映基因表达的动态变化。
ldat <- ReadVelocity(file = "GSM5824332_236D_236D_V300044428.loom")
str(ldat,max.level = 1)
## List of 3
## $ spliced :Formal class 'dgCMatrix' [package "Matrix"] with 6 slots
## $ unspliced:Formal class 'dgCMatrix' [package "Matrix"] with 6 slots
## $ ambiguous:Formal class 'dgCMatrix' [package "Matrix"] with 6 slots
示例数据可以从GEO数据库查询编号:GSM5824332,拉到页面最下方,即可下载到。
2. 数据质量控制
2.1 细胞过滤
过滤掉低质量的细胞是单细胞分析的重要步骤。这里我们保留总表达量大于等于800的细胞。
# 设置过滤阈值,官网给的是1000,但我这个数据设置1000就基本不剩啥了,所以改小了一点。
k <- colSums(ldat$spliced) >= 700;table(k)
## k
## FALSE TRUE
## 7161 908
# 对ldat中的每个矩阵应用相同的过滤
for (i in 1:length(ldat)){
ldat[[i]] <- ldat[[i]][,k]
rownames(ldat[[i]]) <- make.unique(rownames(ldat[[i]]))
}
2.2 创建Seurat对象
将过滤后的数据转换为Seurat对象,便于后续的标准化分析流程。
# 提取spliced和unspliced数据
emat <- ldat$spliced # 已剪接RNA矩阵
nmat <- ldat$unspliced # 未剪接RNA矩阵
scRNA <- as.Seurat(x = ldat)
3. 标准单细胞分析流程
执行Seurat的标准预处理流程,降维聚类分群。
scRNA <- NormalizeData(scRNA)
scRNA <- FindVariableFeatures(scRNA)
scRNA <- ScaleData(scRNA)
scRNA <- RunPCA(scRNA)
scRNA <- FindNeighbors(scRNA, dims = 1:15)
scRNA <- RunUMAP(scRNA, dim = 1:15)
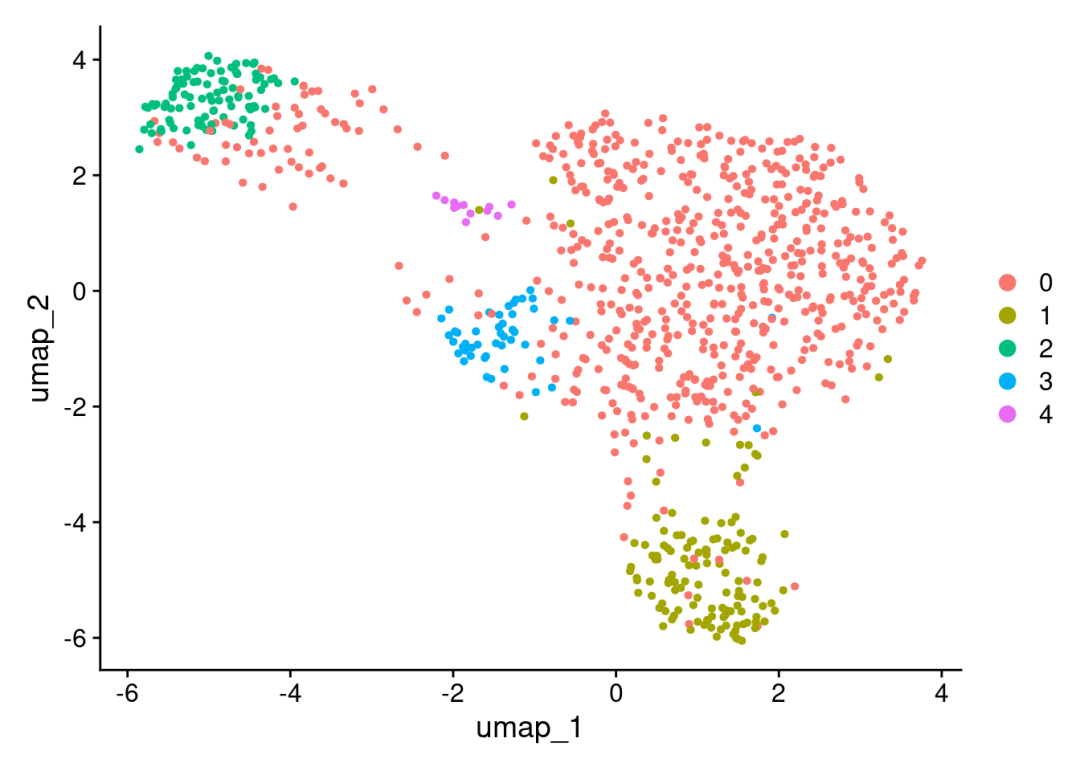
scRNA <- FindClusters(scRNA,resolution = 0.5)
## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 908
## Number of edges: 38414
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.7012
## Number of communities: 5
## Elapsed time: 0 seconds
UMAPPlot(scRNA)
4. 细胞类型注释
4.1 使用SingleR进行自动注释
SingleR是一个基于参考数据集的自动细胞类型注释工具。这里我们使用BlueprintEncode参考数据集。
library(celldex)
library(SingleR)
ls("package:celldex")
## [1] "BlueprintEncodeData" "DatabaseImmuneCellExpressionData"
## [3] "defineTextQuery" "fetchLatestVersion"
## [5] "fetchMetadata" "fetchReference"
## [7] "HumanPrimaryCellAtlasData" "ImmGenData"
## [9] "listReferences" "listVersions"
## [11] "MonacoImmuneData" "MouseRNAseqData"
## [13] "NovershternHematopoieticData" "saveReference"
## [15] "searchReferences" "surveyReferences"
f = "ref_BlueprintEncode.RData"
if(!file.exists(f)){
ref <- celldex::BlueprintEncodeData()
save(ref,file = f)
}
ref <- get(load(f))
library(BiocParallel)
test = scRNA@assays$spliced$data
pred.scRNA <- SingleR(
test = test, # 测试数据
ref = ref, # 参考数据集
labels = ref$label.main, # 使用主要细胞类型标签
clusters = scRNA@active.ident # 按聚类进行注释
)
# 查看注释结果
pred.scRNA$pruned.labels
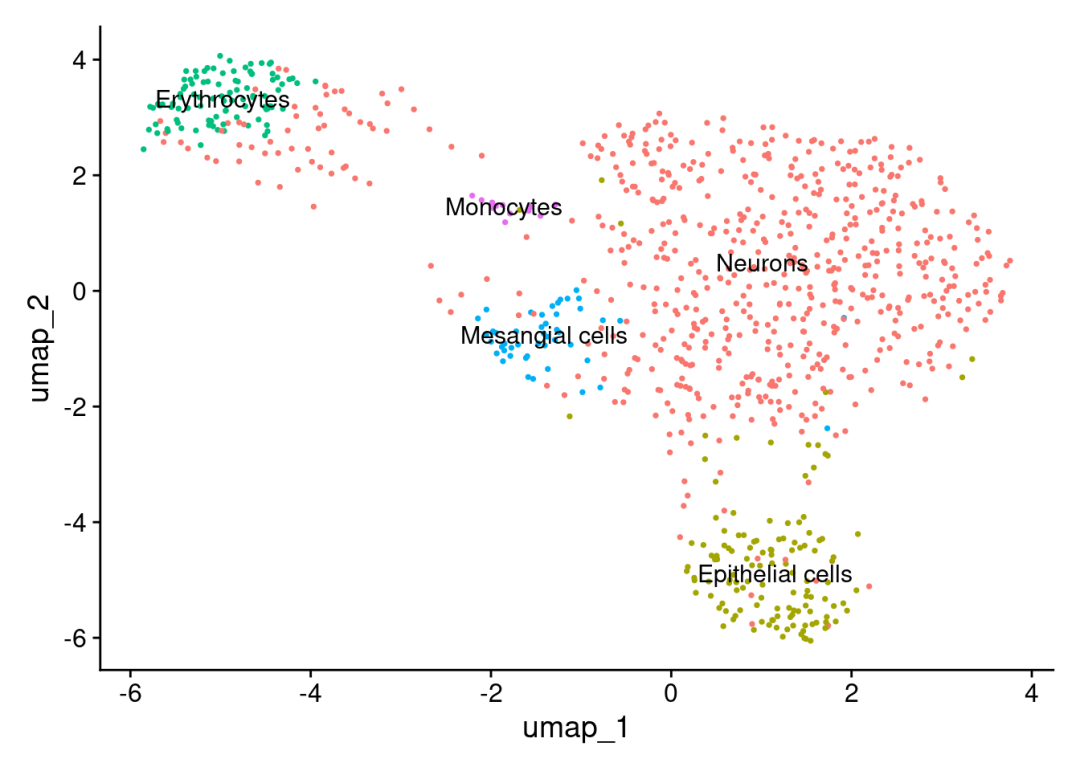
## [1] "Neurons" "Epithelial cells" "Erythrocytes" "Mesangial cells" "Monocytes"
4.2 可视化注释准确性
# 绘制注释得分热图
plotScoreHeatmap(
pred.scRNA,
clusters=pred.scRNA@rownames,
fontsize.row = 9,
show_colnames = T
)

4.3 更新细胞类型标签
将自动注释的结果应用到Seurat对象中。
# 创建新的聚类标签
new.cluster.ids <- pred.scRNA$pruned.labels
names(new.cluster.ids) <- levels(scRNA)
# 查看原始聚类
levels(scRNA)
## [1] "0" "1" "2" "3" "4"
# 重命名聚类
scRNA <- RenameIdents(scRNA,new.cluster.ids)
# 查看更新后的聚类
levels(scRNA)
## [1] "Neurons" "Epithelial cells" "Erythrocytes" "Mesangial cells" "Monocytes"
# 绘制UMAP图
p3 <- DimPlot(
scRNA,
reduction = "umap",
label = T, # 显示标签
pt.size = 0.5 # 点的大小
) + NoLegend()
p3
5. RNA速率分析
5.1 准备速率分析所需数据
为RNA速率分析准备必要的数据,包括细胞分组信息、颜色设置和细胞间距离计算。
# 设置细胞群颜色
library(paletteer)
n = nlevels(scRNA) # 获取细胞类型数量
library(RColorBrewer)
# 使用调色板为不同细胞类型分配颜色
cell.colors <- colorRampPalette(paletteer_d("RColorBrewer::Set1"))(n)[Idents(scRNA)]
names(cell.colors) = colnames(scRNA)
5.2 计算RNA速率
使用gene.relative.velocity.estimates函数计算每个基因的相对速率。
scRNA = RunVelocity(object = scRNA, deltaT = 1, kCells = 25, fit.quantile = 0.02)
6. 速率可视化
6.1 全局速率场可视化
在UMAP图上展示整体的RNA速率场,箭头表示细胞状态转换的方向。
# 获取UMAP坐标
emb <- scRNA@reductions$umap@cell.embeddings
# 绘制全局速率图
show.velocity.on.embedding.cor(
emb, # UMAP坐标
Tool(object = scRNA,slot = "RunVelocity"), # 速率信息
cell.colors=cell.colors, # 细胞颜色
n=100, # 显示的箭头数量
scale = 'sqrt', # 箭头缩放方式
cex =1.2, # 点的大小
arrow.scale = 1, # 箭头大小缩放因子
show.grid.flow = TRUE, # 是否显示网格流
min.grid.cell.mass = 0.5, # 最小网格细胞质量
grid.n = 20, # 网格数量
arrow.lwd = 1, # 箭头线宽
do.par = T, # 是否设置图形参数
cell.border.alpha = 0.5 # 细胞边界透明度
)
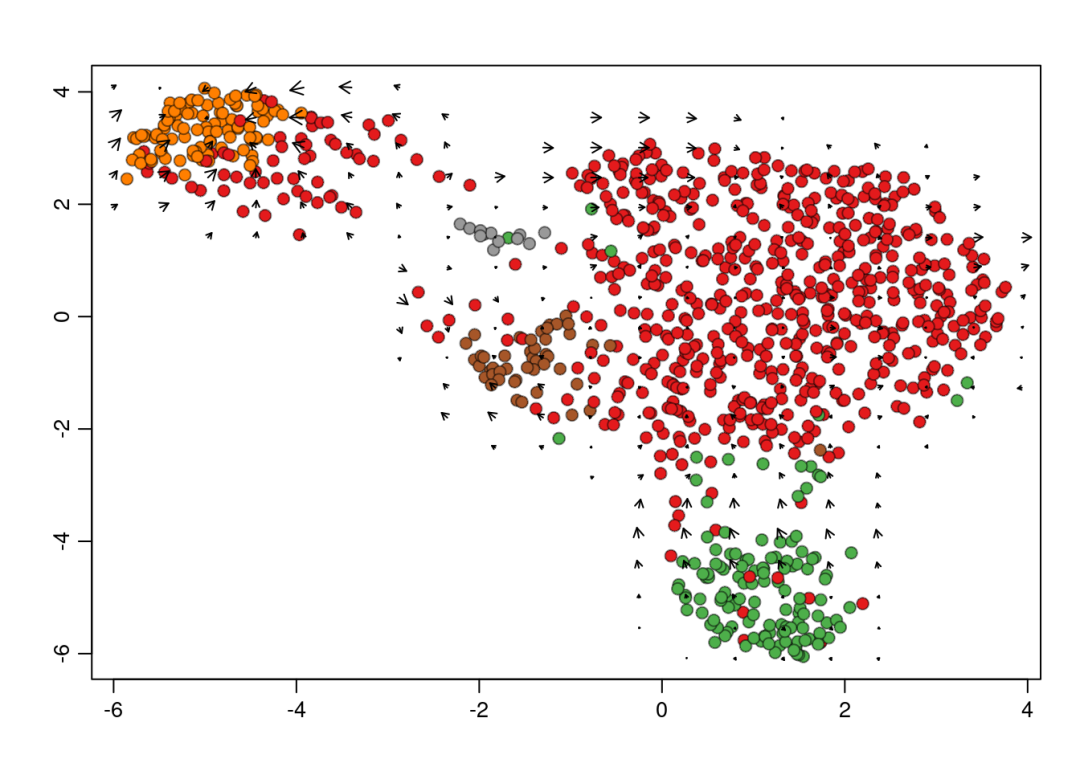
## delta projections ... sqrt knn ... transition probs ... done
## calculating arrows ... done
## grid estimates ... grid.sd= 0.3741385 min.arrow.size= 0.007482771 max.grid.arrow.length= 0.1072256 done
箭头方向:指向细胞即将分化成的未来状态。
颜色:按照细胞类型分配颜色。
发育方向:沿箭头从起始细胞群流向目标细胞群即可判断分化路径。
6.2 特定基因速率可视化
查看单个基因的表达动态,了解该基因在不同细胞状态间的表达变化趋势。
# 选择要查看的基因
gene <- "UBA52"
cell.dist <- as.dist(1 - armaCor(t(scRNA@reductions$pca@cell.embeddings)))
# 绘制特定基因的速率图
gene.relative.velocity.estimates(
emat, # spliced矩阵
nmat, # unspliced矩阵
deltaT=1, # 时间步长
kCells = 20, # 最近邻细胞数
kGenes=1, # 基因数
fit.quantile=0.02, # 拟合分位数
cell.emb=emb, # 细胞嵌入坐标
cell.colors=cell.colors, # 细胞颜色
cell.dist=cell.dist, # 细胞距离
show.gene=gene, # 要显示的基因
old.fit= Tool(object = scRNA,slot = "RunVelocity"), # 之前的速率拟合结果
do.par=T # 是否设置图形参数
)
## calculating convolved matrices ... done
## [1] 1
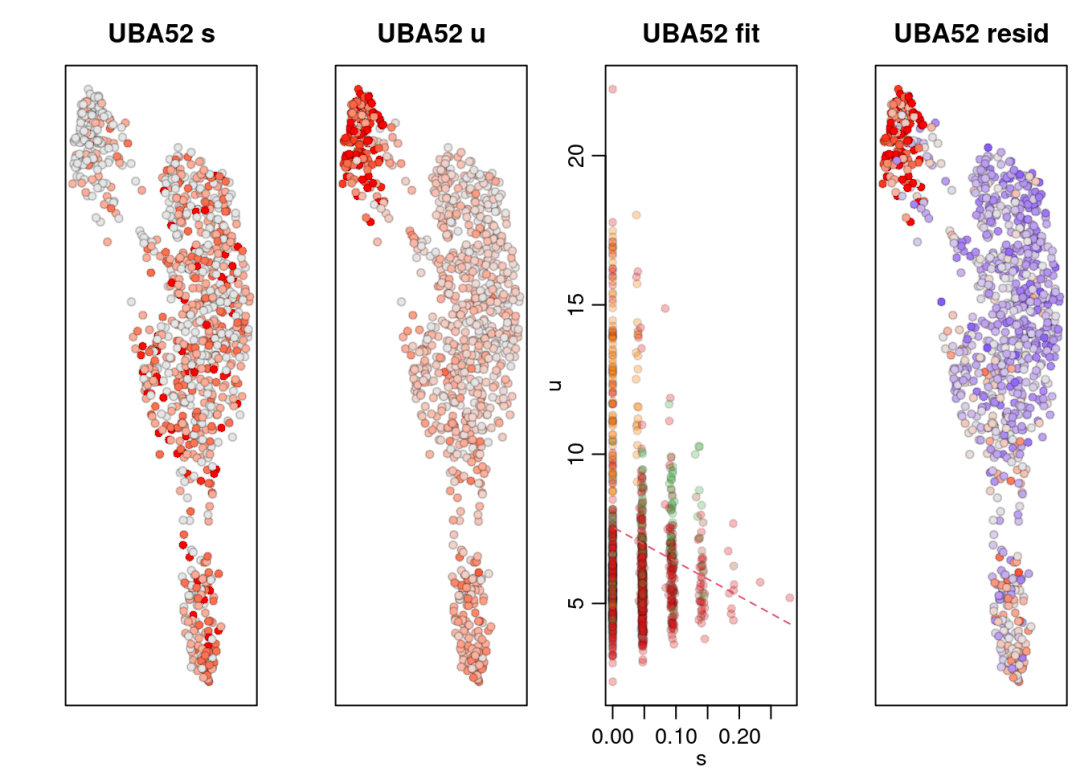
图1:s(spliced):已剪接的基因在细胞中的表达情况。颜色深浅表示表达量的高低,颜色越深,表示表达量越高。
图2:u(unspliced):显示了未剪接的基因在细胞中的表达情况,颜色深浅表示表达量的高低。
图3:fit:已剪接和未剪接基因表达量之间的关系。颜色代表细胞类型。
红色的虚线代表模型拟合的最佳线性关系。
位于拟合线上方的点:u > 模型预测的值,可能表明这些细胞中基因转录正在增加。
位于拟合线下方的点:u < 模型预测的值,可能表明这些细胞中基因转录正在减少。
图4:resid:基因表达的残差,即实际观测值与模型预测值之间的差异。有助于识别细胞中基因表达的异常变化,颜色越深表示残差的绝对值越大。
残差为正说明u<模型预测值,基因表达可能正在增加。
残差为负说明u>模型预测值,基因表达可能正在减少。
如果再详细一点的话...
残差为正:这表明在模型预测的未剪接RNA量相对于实际观测到的量来说偏低。这可能是因为模型没有完全捕捉到基因表达的动态变化,或者实际上基因表达的速率确实增加了。在这种情况下,细胞可能正在增加特定基因的转录,导致更多的未剪接RNA积累,因为新的转录本正在被合成,但还没有足够的时间进行剪接。因此,正残差通常被解释为基因表达可能正在增加的信号。
残差为负:这表明在模型预测的未剪接RNA量相对于实际观测到的量来说偏高。这可能意味着基因表达的速率实际上正在减少,导致较少的未剪接RNA被合成,或者剪接过程比模型预测的要快,使得未剪接RNA迅速转化为已剪接RNA。在这种情况下,负残差通常被解释为基因表达可能正在减少的信号,或者剪接过程加速,使得未剪接RNA的量低于预期。
7.遇到的报错及其解决办法
第一个是:
Error in seq.default(rx[1], rx[2], length.out = grid.n) : 'from' must be a finite number
解决办法是修改源代码,加上NA处理机制,详见:https://www.jianshu.com/p/96a86748f593
修改之后的文件我也提供一份。请在"生信星球"聊天框回复velo1054,即可获得。
第二个:
BLAS : Program is Terminated. Because you tried to allocate too many memory regions.
解决办法是开头那句代码:
RhpcBLASctl::blas_set_num_threads(1)
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-09-15,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号