基于bulk miRNA数据和参考库预测细胞组成类型比例的去卷积方法
原创
基于bulk miRNA数据和参考库预测细胞组成类型比例的去卷积方法
原创
元莘生物
发布于 2025-11-03 15:23:24
发布于 2025-11-03 15:23:24
复旦大学智能医学研究院及附属浦东医院杨桢青年副研究员为该论文唯一通讯作者,复旦大学生物医学研究院硕士研究生朱韶英及皖南医学院弋矶山医院杨辉博士为该论文共同第一作者。该工作得到中国科学院上海营养与健康研究所Andrew E. Teschendorff教授的合作支持。同时该研究得到了国家基金委、上海市教委人工智能赋能学科跃升计划项目的资助。
摘要
miRNA在生物发育和疾病进展中扮演着至关重要的角色,并可作为重要的生物标志物。同时miRNA具有高度的细胞表达异质性。目前单细胞转录组测序技术在miRNA表达定量方面仍无法有效展开,且大多数miRNA图谱样本缺乏与之匹配的可用于细胞类型去卷积的mRNA表达或DNA甲基化数据;因此,开发一种能够估算复杂组织中miRNA数据中的细胞类型比例的计算方法至关重要。
本文中构建了一种创新性的miRNA表达参考文库和去卷积工具DeconmiR,用于评估复杂组织中的细胞类型构成;该工具在全血及不同实体组织中的去卷积分析均表现出高准确性和稳健性。通过将此工具应用于多种生物学情景,研究验证了其在筛选年龄相关miRNA、检测新冠等传染病的免疫微环境,以及鉴定用于癌症早期诊断和预后评估的细胞类型特异性miRNA生物标志物等方面的应用价值。本项研究工作为miRNA数据的细胞类型混合物精准去卷积分析建立了一个计算框架。
引言
miRNA在多细胞生物体的细胞周期、增殖、分化、发育和凋亡等广泛的生理过程中发挥重要作用。而且大部分miRNA存在高度的细胞异质性,在临床研究的组织miRNA数据中,观察到了miRNA在不同组织中的较大差别,表明其有作为预测、诊断或预后标志物的能力。现阶段,公开数据库中存在大量的miRNA分子谱数据,通过对这些公开数据的探索,发现了越来越多的miRNA生物标志物;但是这些谱数据多是来自细胞混合的“bulk”检测,细胞异质性的数据因素已经混入其中,即观察到的miRNA差异可能是由于组织中的特异细胞类型中表达的改变。另一方面,细胞类型的相对组成变化在各种生理过程中起着重要作用而且细胞类型量化也可解释许多疾病的成因和演化(例如癌症肿瘤微环境的动态变化),因此从这些miRNA表达谱数据中解析细胞类型和组成对于鉴定miRNA疾病标志物用着重要的作用。
单细胞转录组测序暂无法有效鉴定单细胞级别的miRNA表达数据,可以替代的方法是通过计算机模拟细胞类型去卷积来进行细胞类型组成鉴定;目前bulk数据中mRNA数据和甲基化数据可以通过去卷积算法有效解析复杂组织的细胞类型组成。算法的运行逻辑是:基于不同组织的不同细胞类型的特定标志物的先验参考谱信息(分选的细胞类型或更精细的单细胞谱数据),例如CIBERSORT及其后继者CIBERSORTx利用支持向量回归方法和从纯化的细胞群体或单细胞数据生成的参考用于细胞组成估计;但公共数据中的miRNA数据大多数没有对应匹配的先验参考谱信息(例如GEO中有11万个以上的miRNA表达谱数据,但其中只有约5%具有匹配的先验参考谱信息);因此,本研究中从分选的细胞中构建了miRNA参考矩阵,并开发了去卷积方法DeconmiR,DeconmiR可以从混合样品的miRNA谱中解析不同细胞类型的相对比例,并从各种实验和临床样品中评估了上述方法(患者预后、年龄相关,感染新冠的免疫景观,肺癌特异性细胞类型),均具有很高的预测准确率;综上,本研究设计了一个高效的去卷积方法,可以通过bulk miRNA谱数据推断组织中细胞类型的相对比例。
研究思路
本研究的核心在于构建高质量的miRNA表达参考文库(细胞亚群级别)并开发配套的去卷积工具DeconmiR,以通过bulk miRNA数据预测复杂组织中的细胞类型和比例。研究团队通过公开数据收集组织分选出纯化细胞的miRNA数据,构建针对血液和多种实体组织的特异性表达特征谱,然后开发了去卷积工具DeconmiR,并通过计算机模拟混合样本和真实临床样本进行双重验证,最终证实了该方法DeconmiR的精确性和有效性,其可以用于生物标志物的鉴定。
研究结果
血液样本miRNA表达矩阵的构建
参考依赖性去卷积方法完全依赖于细胞类型特异性标志物的文库;参考文库应由不同细胞类型中每种细胞特异性高表达的miRNA组成;研究收集了257个外周血miRNA数据(47个个体中分选的6种分选的细胞亚型的数据),从中筛选了纯度较高的197个样品数据用于参考构建:limma鉴定差异miRNA--tSNE聚类--构建参考矩阵。上述鉴定过程中,鉴定到部分特异性的miRNA:miR-155:淋巴细胞中高表达;miR-181:人造血祖细胞向NK细胞分化中高表达。细胞特异性miRNA靶向基因的GO富集分析也揭示特定细胞类型的功能特异性。
本研究设计的DeconmiR方法,用于通过使用稳健偏相关(RPC)进行基于miRNA分析的细胞类型去卷积,其有效性已通过应用于作者先前基于DNA甲基化的去卷积的工作得到证明。首先使用上述具有已知免疫细胞比例的的257个miRNA数据进行评估,数据分析显示预测结果和FACS估计的细胞比例之间具有高度一致性,对于六种细胞类型,平均均方根误差(RMSE)为0.11。为了进一步评估DeconmiR是否可以通过获取细胞混合的miRNA谱数据来评估细胞相对比例,研究通过随机生成不同免疫细胞混合物比例的模拟组织(混合比例已知),生成了100种混合物,针对混合物的miRNA数据进行分析显示,DeconmiR在所有细胞类型中获得了近乎完美的R2值,RMSE范围为1%至3.2%,证实了我们的方法可以通过使用参考文库准确地定量血细胞类型比例。
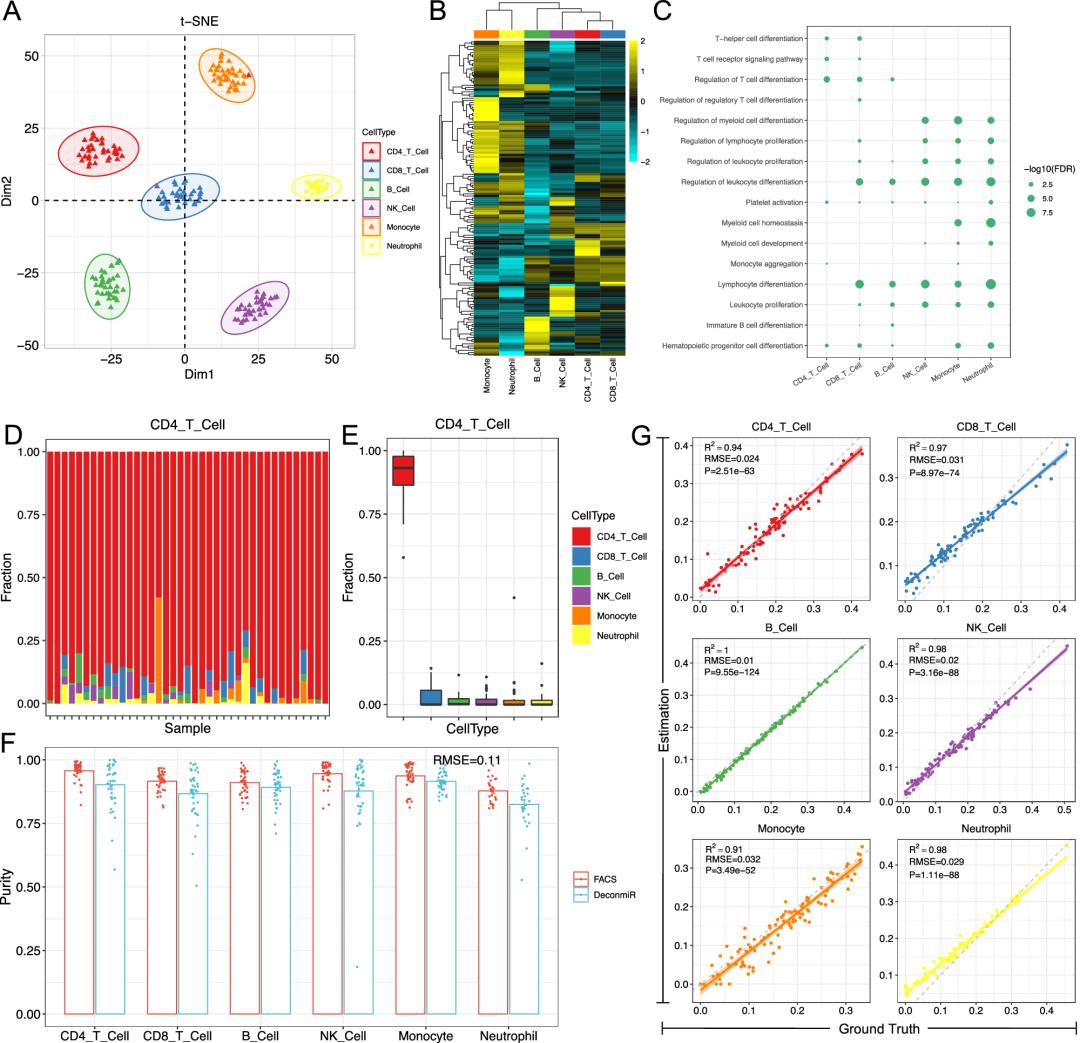
DeconmiR方法优势
- 捕获稀有细胞组分:研究基于单个细胞类型不同比例的的混合物数据进行了去卷积,通过设置比例梯度(1%、3%、5%、7%和9%),并随机分配其他组分的比例,生成了100份计算机模拟混合物样品。结果表明,DeconmiR的预测接近于实际情况,进一步说明了DeconmiR在推断稀有细胞组成方面的稳健性。
- 独立数据验证:14个已知细胞比例的混合细胞样品的miRNA数据,验证数据集显示,DeconmiR方法实现了高度准确的细胞类型分数估计
- 数据方法优势:研究通过将DeconmiR与众多优秀去卷积方法(SLE、NNLS、CIBERSORT、DeconRNAseq(QP))进行比较,在同样的参考库基础上,DeconmiR对细胞类型识别的表现优于其他方法(除了单核细胞中,QP表现略好)。
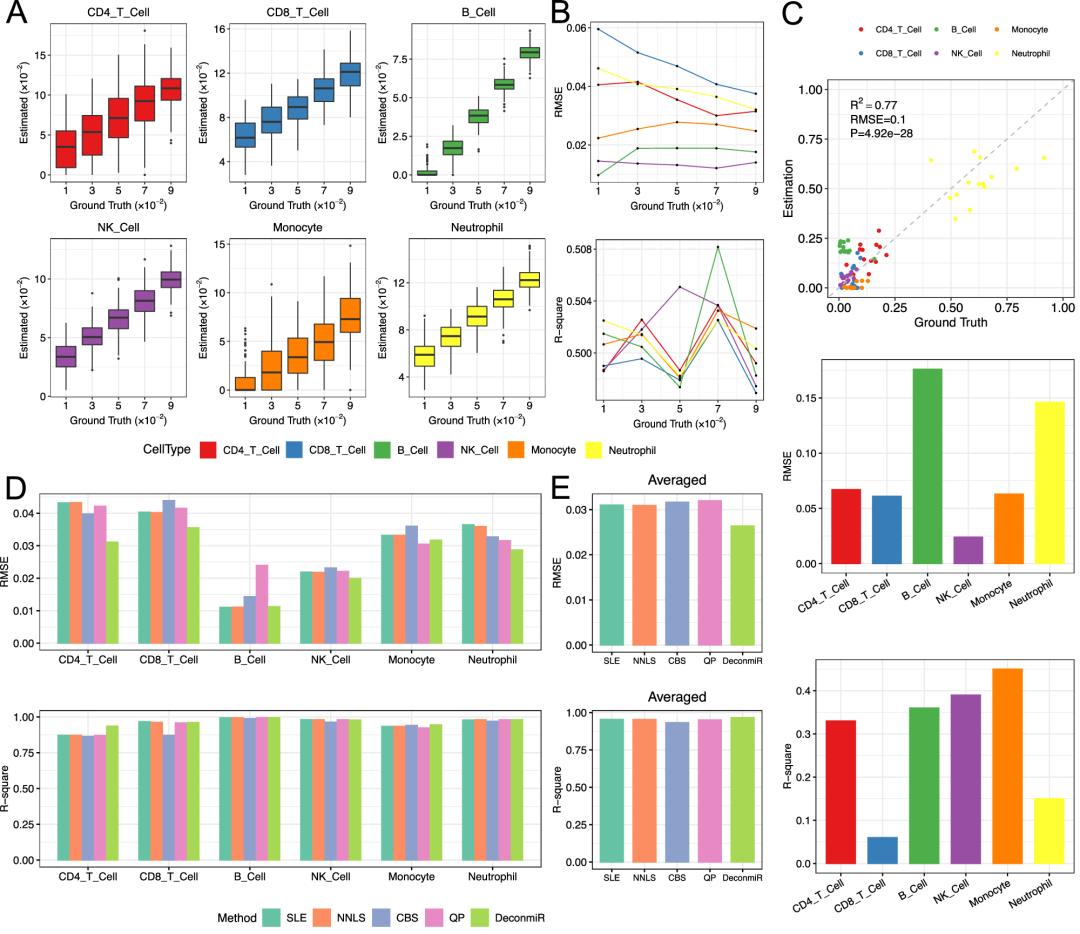
DeconmiR应用优势
通过数据验证DeconmiR对急性髓系白血病的潜在诊断价值、鉴定衰老相关的miRNA的敏感性、鉴定新冠患者血液样本的细胞类型组成变化、揭示乳腺癌种的免疫细胞浸润中均表现出算法的有效性、跨肿瘤类型的细胞类型、吸烟者肺癌上皮细胞的miRNA变化;在构建了特定组织的参考矩阵的基础上(获取细胞特异性miRNA),通过构建好的方法预测特定组织的细胞类型,上述几个案例的结果都证明了DeconmiR用于实体组织和血液细胞比例估计的可靠性。
展示了DeconmiR在不同应用环境中的有效性。通过将我们的方法应用于人类AML恶性肿瘤,DeconmiR表明疾病和对照样品之间不同细胞类型组成的差异。通过我们的方法预测的细胞分数能够区分样本到亚组,这表明AML的临床诊断和预后的潜在价值。当应用于年龄相关的miRNA谱研究时,与未调整的细胞类型分析相比,它可以识别出两倍以上的年龄相关的miRNA,证实DeconmiR可以显着提高筛选灵敏度。此外,它成功捕捉了COVID-19患者血液中主要细胞类型的动态变化,这在探索感染性疾病的免疫反应方面显示了重要的应用价值。
我们还将基于参考的框架的成功扩展到血液以外的复杂组织。在这样做时,我们不仅使用几个独立的数据集为各个组织类型提供详细的miRNA参考数据库,而且还提供通用参考,其能够通过使用分层去卷积策略40可靠地估计复杂组织中上皮、免疫和基质(主要是成纤维细胞)细胞的分数。使用我们的新的参考矩阵,我们证明了复杂的组织表现出广泛的免疫细胞污染。以乳腺组织为例,我们的分析表明,参考矩阵不仅可以用于估计样本的细胞含量,而且可以通过控制灵敏度和特异性来推断上皮区室中发生的癌症特异性miRNA生物标志物。在肺癌的情况下,我们表明,许多排名靠前的吸烟相关的miRNA检测到的分析进行支气管气道可以检测到特异性的肺癌样本的上皮隔室,而许多其他的也发生在免疫隔室。支气管和肺上皮中这些细胞类型特异性miRNA的变化可能影响或反映了导致肺癌发展的调节网络的失调。
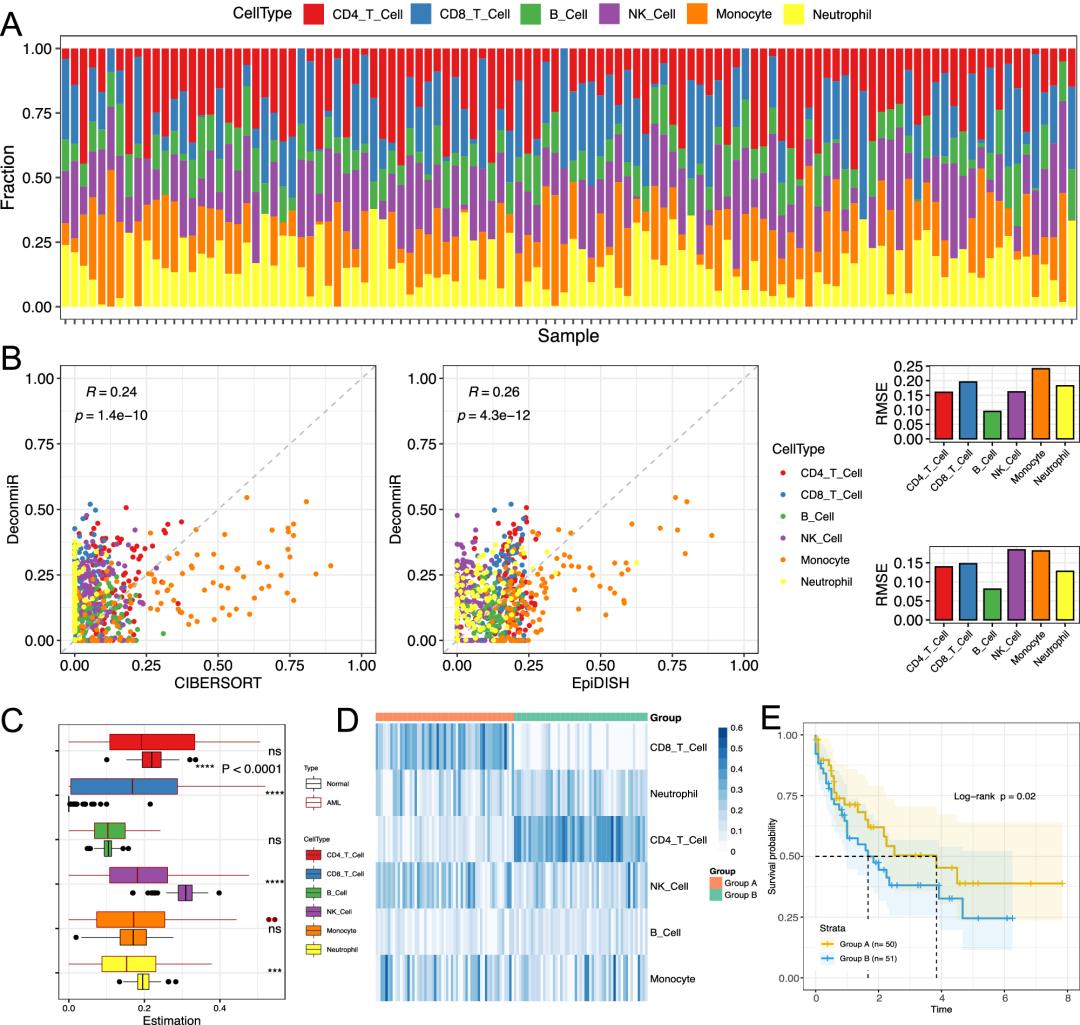
结果讨论
通过去卷积算法计算细胞类型是解析复杂组织的异质性的有效策略,本研究开发了DeconmiR方法,可以通过采用miRNA表达谱数据来预测细胞类型组成,通过一系列的模式和实验数据,研究证明了DeconmiR可以对各种细胞类型进行高精度预测,同时对数据噪声具有鲁棒性。为了保证方法对复杂组织的细胞类型定量的性能,DeconmiR利用几种方法来获得高度细胞特异性表达的miRNA,并构建最能提供去卷积信息的参考特征矩阵;研究结果表明,对于小于200个miRNA的文库(远小于基于转录组的去卷积方法所招募的基因),可以实现良好的细胞类型区分。根据研究中的经验,当对miRNA表达谱进行去卷积时,可以在对数标度中实现更准确的去卷积。
文章还展示了DeconmiR在不同应用环境中的有效性。这些结果对未来的miRNA谱研究具有深远的意义,随着更多的工作证明miRNA谱数据在这一领域的价值,它可以帮助改善生物学揭示,优先考虑需要功能验证的候选人,并阐明不同疾病的因果途径。研究还指出方法的应用边界和局限性:例如一些特征非常相似的细胞类型,会导致去卷积过程中的混淆;还有高度相关的miRNA(来源相同或有相同的seed区域)将冗余信息引入参考文库,从而导致特定细胞类型的权重过高。
综上,研究提出了一种基于参考文库的去卷积算法DeconmiR,可以用于预测bulk miRNA数据中的细胞组成比例。
引用该文献
Zhu, Shaoying, et al. “An Improved Reference Library and Method for Accurate Cell-Type Deconvolution of Bulk-Tissue miRNA Data.” Nature Communications, vol. 16, no. 1, Jul. 2025, p. 5508. www.nature.com, https://doi.org/10.1038/s41467-025-60521-x.
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号