day9-10
原创
day9-10
原创
用户11775359
发布于 2025-08-17 22:34:27
发布于 2025-08-17 22:34:27
day9
背景知识:
GPL文件学习:
Data table header descriptions:是芯片注释平台的表头描述
ID:是指探针的编号
probeset_id:表达集的探针编号
seqname:染色体名称
strand:是指正列还是负列
start:起始位置
stop:终止位置
total_probes:总的探针数目
gene_assignment:基因的描述
mrna_assignment:mrna的描述
swissprot:蛋白数据库的描述
category:种类
spot_ID:spot格式的ID
箱线图检查芯片数据,如果数据不齐::normalize between array
当没有GPL 包的时候可以使用方法二 :网站下站GPL.txt文件自己提取ID和symbol
day 10
一、差异基因聚类图 火山图
1、聚类和分组的爱恨情仇:当聚类和分组呈现类似田字格时,说明选择的基因比较有代表性
2、不好分组的聚类分析结果 可以放在文章里吗?不建议 因为审稿人会问 本质是选择的基因不太争气,处理方式可以取消热图聚类
聚类分析取消列的聚类后,可以单独把两组分别放在一次,参考洁笔记:https://www.jianshu.com/p/b94449be175a
富集分析需要symbol转换为entrezid,每种物种的使用的包不同
library(clusterProfiler)
library(org.Hs.eg.db)
s2e = bitr(deg$symbol,
fromType = "SYMBOL",
toType = "ENTREZID",
OrgDb = org.Hs.eg.db)#⭐Hs是人类,注意物种
#一部分基因没匹配上是正常的。只要物种正确,<30%的失败都没事。
#其他物种http://bioconductor.org/packages/release/BiocViews.html#___OrgDb
#⭐检查行数,减少太多说明不正常
##结果行数增加和减少都是正常的二、富集分析
一个数据库中有多个基因集
一个基因集中有多个基因
kegg的通路和GO里的term都是基因集
rm(list = ls())
load(file = 'step4output.Rdata')
library(clusterProfiler)
library(ggthemes)
library(org.Hs.eg.db)
library(dplyr)
library(ggplot2)
library(stringr)
library(enrichplot)
#(1)输入数据
gene_diff = deg$ENTREZID[deg$change != "stable"]
#(2)富集
#⭐下面的三句都要注意物种
ekk <- enrichKEGG(gene = gene_diff,organism = 'hsa')
#其他物种https://www.genome.jp/kegg/catalog/org_list.html
ekk <- setReadable(ekk,OrgDb = org.Hs.eg.db,keyType = "ENTREZID")
#如果ekk是空的,这句就会报错,因为没富集到任何通路。
ego <- enrichGO(gene = gene_diff,OrgDb= org.Hs.eg.db,
ont = "ALL",readable = TRUE)
#setReadable和readable = TRUE都是把富集结果表格里的基因名称转为symbol
class(ekk)
#(3)可视化
#barplot可以换成dotplot就改成气泡图了
barplot(ego, split = "ONTOLOGY") +
facet_grid(ONTOLOGY ~ ., space = "free_y",scales = "free_y")
barplot(ekk)
#如果ekk中没有padj<0.05的通路,就会报错,因为只画padj<0.05,没有参数
# 更多资料---
# GSEA:https://www.yuque.com/docs/share/a67a180f-dd2b-4f6f-96c2-68a4b86fe862?#
# Y叔的书:http://yulab-smu.top/clusterProfiler-book/index.html
# GOplot:https://mp.weixin.qq.com/s/LonwdDhDn8iFUfxqSJ2Wew
# 网上的资料和宝藏无穷无尽,学好R语言慢慢发掘~富集结果解读:
第一列:通路名
description 通路的描述
p值 矫正p q值:富集结果是够显著
geneID 同时是差异基因和通路里面的基因
count:同理里有几个基因
GeneRatio: 通路中差异基因个数/差异基因中有多少被数据库收录
BgRatio 通路中有多少基因/数据库中所有通路总共有多少基因
可视化
柱状图barplot和气泡图dotplot
富集不到的补救措施
火山图横坐标log FC 范围一般是-13~13 太大可能没取log;太小可能多取了log ,或者数据本身不争气
part4-复杂数据及分析(都在GEO_learnmore 文件夹里 以后慢慢学吧)
1.多分组
2.多数据联合分析
1) 各自差异分析 基因取交集
或者
2) 先合并,后差异分析:
需要处理批次效应 Batch effect
不能选择一个数据全是处理组;一个全是对照组多数据合并;
函数:limma::removeBarchEffect(); sva::ComBat
3.加权共表达WGCNA
1)要求至少15样本
2)图1.软阈值图需要放进文章
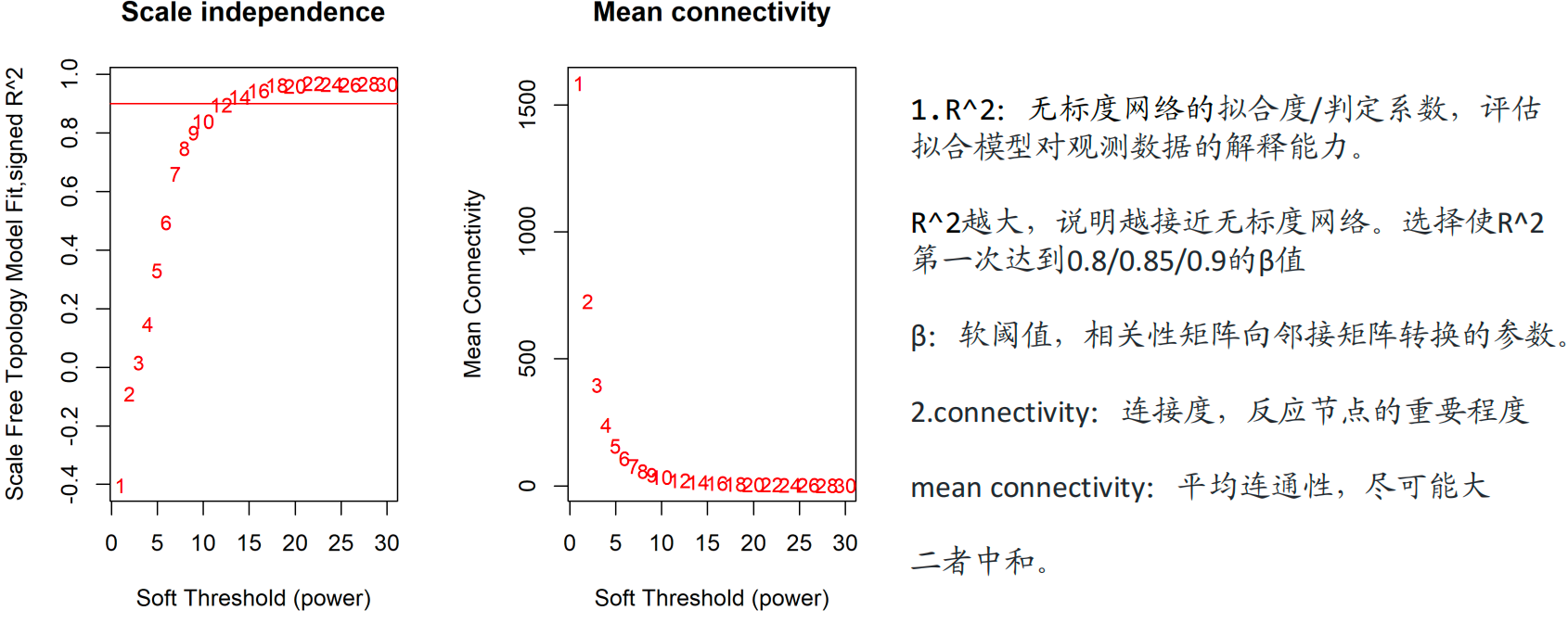
背景知识:基因的网络应该满足无标度网络
3)图2.基因模块化(对基因进行聚类)
同一模块的基因通常有相似的功能
灰色的模块聚类失败 没有背景知识分组表型等可概括
图3. 相关系数都是比较小数字 小于0.5,相关性小, 就不建议 WGCNA这种方法挑选基因
4.蛋白互作网络
B 站视频 cytoscape
ppt里面有具体操作步骤
5.网络药理学

需要进一步学习
有效成分——预测靶基因
后面有链接可以去学
批次效应可以通过PCA图和箱线图来识别,有就要去批次效应
摆脱弱者思维 对自己进行积极的心里暗示,多找客观原因
引用至生信技能树 小洁老师
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号