day 7-8 GEO数据挖掘
原创
day 7-8 GEO数据挖掘
原创
用户11775359
修改于 2025-08-15 13:40:21
修改于 2025-08-15 13:40:21
1.常见图表讲解
1.1背景:
广义基因有6w+ 狭义2w+ 共12种类
表达矩阵:不同样本的检测基因表达量
数据从哪里来:GEO NHANES(临床) TCGA ICGC CCLE SEER(临床
)
有什么类型数据?
1/基因表达芯片- 光信号值2/转录组bulk转录组- count矩阵 3/单细胞转录组 - count矩阵 4/突变、甲基化、拷贝数变异 5/空间转录组
1.2怎样筛选基因
数据下载-数据整理-差异分析(芯片和转录组统计学原理和工具不同)- WGCNA(加权共表达网络)-富集分析(给差异基因找归宿 GO/DO/ KEGG/ GSEA msigdb)-PPI(蛋白蛋白互作网络,不需要R语言)-预后分析(影响生存的疾病)
下载包的经验:1/换镜像 2/换网络 3/确认包的来源途径(CRAN/Biocondutor···) 批量安装包的代码在pipeline-00_pre_install.R
R语言软件版本可能和镜像有关联,注意及时升级R
不鼓励官网下载包手动安装的方式
1.3 常见的图
1、热图:有聚类和基因上调下调的信息
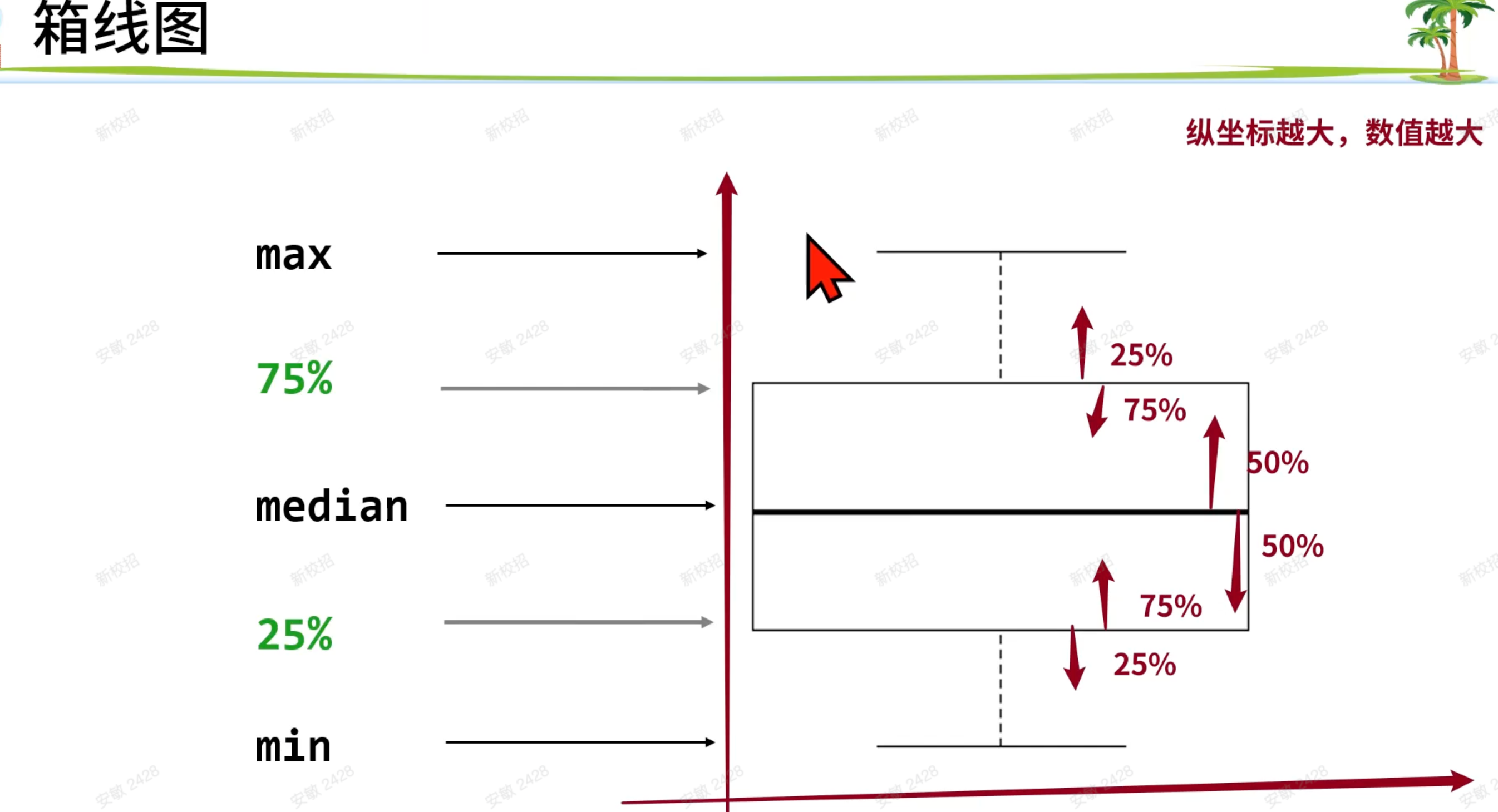
2、散点图和箱线图:
箱线图是散点图的精华
箱线图输入数据是一个连续型数据和一个有重复值的离散值向量
上到下五条线对应:
最大值 75% 中位数 25% 最小值
线段外侧为离群值
散点图就箱线图均用于组件比较
3、多基因差异分析—>火山图(含有log FC 和p值)
重点: 芯片分析的起点是一个取过log的表达矩阵(0~20)
如果拿到的是未log(0~很大),需要自行log
1)注意:logFC的值一般在0~20,看到几千几万的一定有问题(忘记取logFC)
2)log FC 确定上下调整,p值决定是否显著(0.05,0.01)
3) logFC 常见阈值「至少大于0.58(是1.5倍),常为1(是2倍)」
4)p值越小,- log10(p)越大,越有信心认为差异显著
5) 左上角及右上角为最有差异的基因
在火山图上标记差异基因:https://cloud.tencent.com/developer/article/1486128
4、主成分分析(PCA)
4.1通过该分析图片表达的信息为:组内重复是否号,组件差别是否大
主成分:为多个旧变量组合的新变量
原本十几个变量,现在可能只需要2~3个主成分就能代表大部分信息,而且这些主成分之间互不相关!
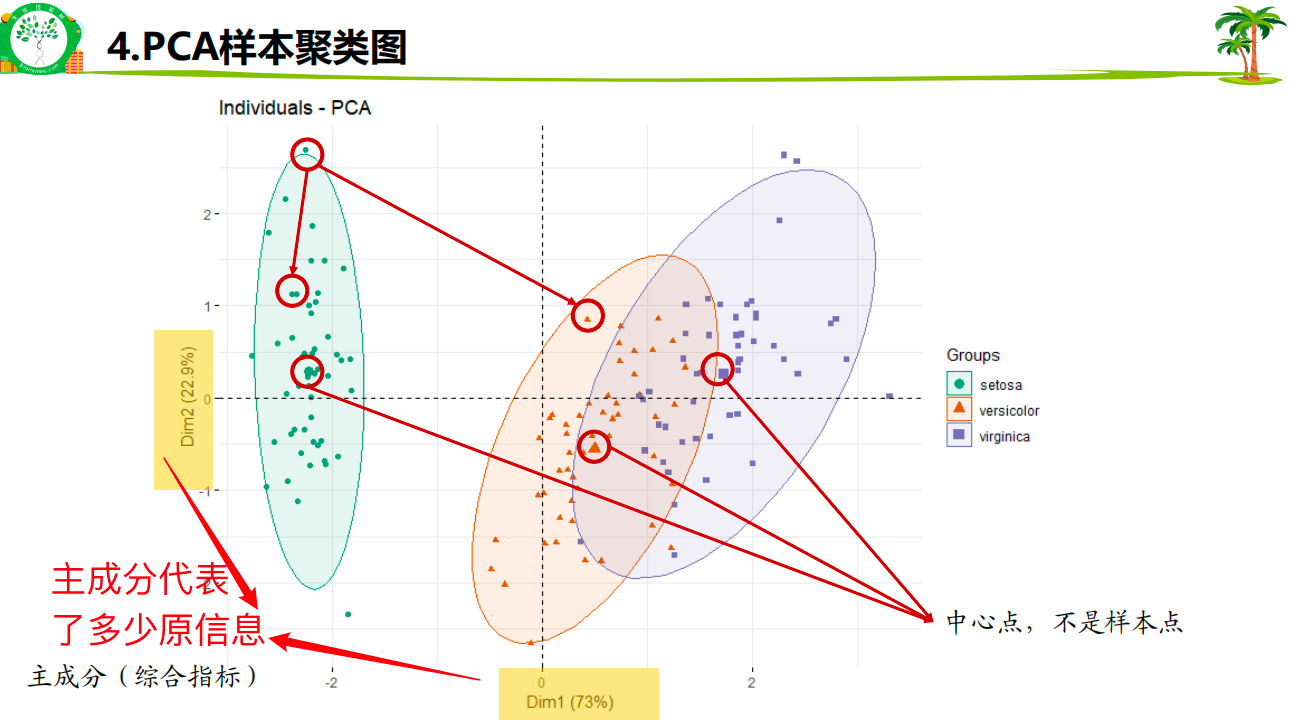
图上的点代表样本,点与点距离代表差别
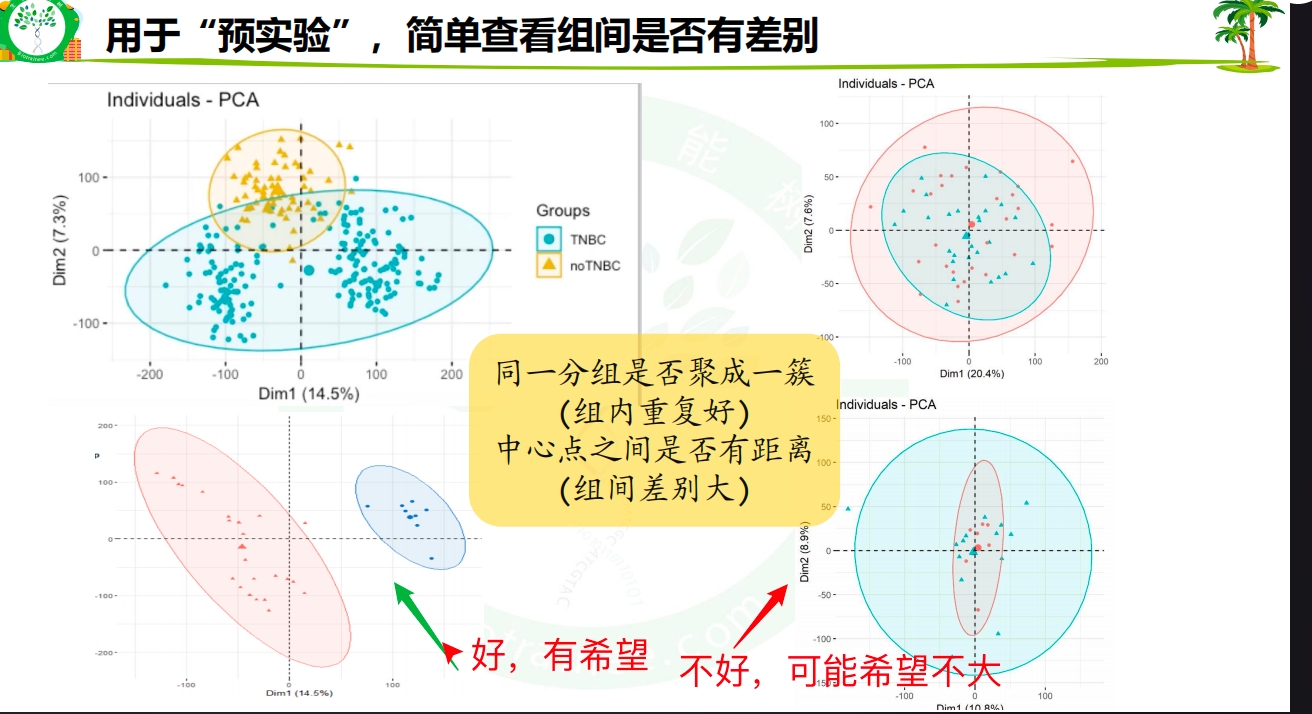
4.2 用途:用于“预试验”,简单查看组间差别
刚得知小洁老师2018年才学R语言,现在如此精通,不过老师做笔记的习惯真的非常好,已经产生超级多笔记了。
2.GEO背景介绍+分析思路
表达数据实验设计:分组需要有意义
分组为病变组织VS 健康组织
如果公共数据库没有,需要自己测
2.1数据挖掘:有差异的材料→差异基因→找功能/找关联→解释差异,缩小基因范围
下载数据下载GSE
2.2分析思路:
2.2.1分析流程:
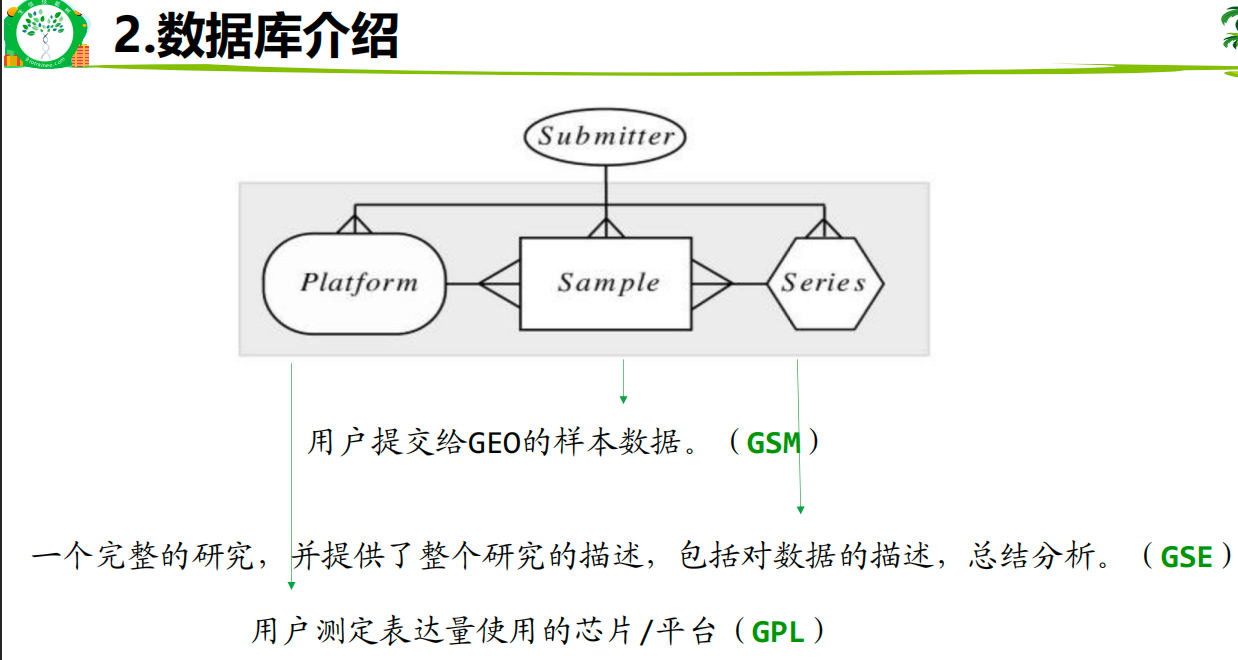
找数据-下载并读取数据-表达矩阵+临床分组信息- GPL编号(探针注释:探针和基因之间的对应关系)——数据探索(分组间是否有差异:PCA/最离散的一些基因的热图)——差异分析和可视化(火山图/差异基因热图)
探针和基因之间的对应关系自己找,不通过GEO下载:因为基因注释每隔几个月就会更新
2.2.2 别人文章用过的数据还能用
2.2.3 表达矩阵
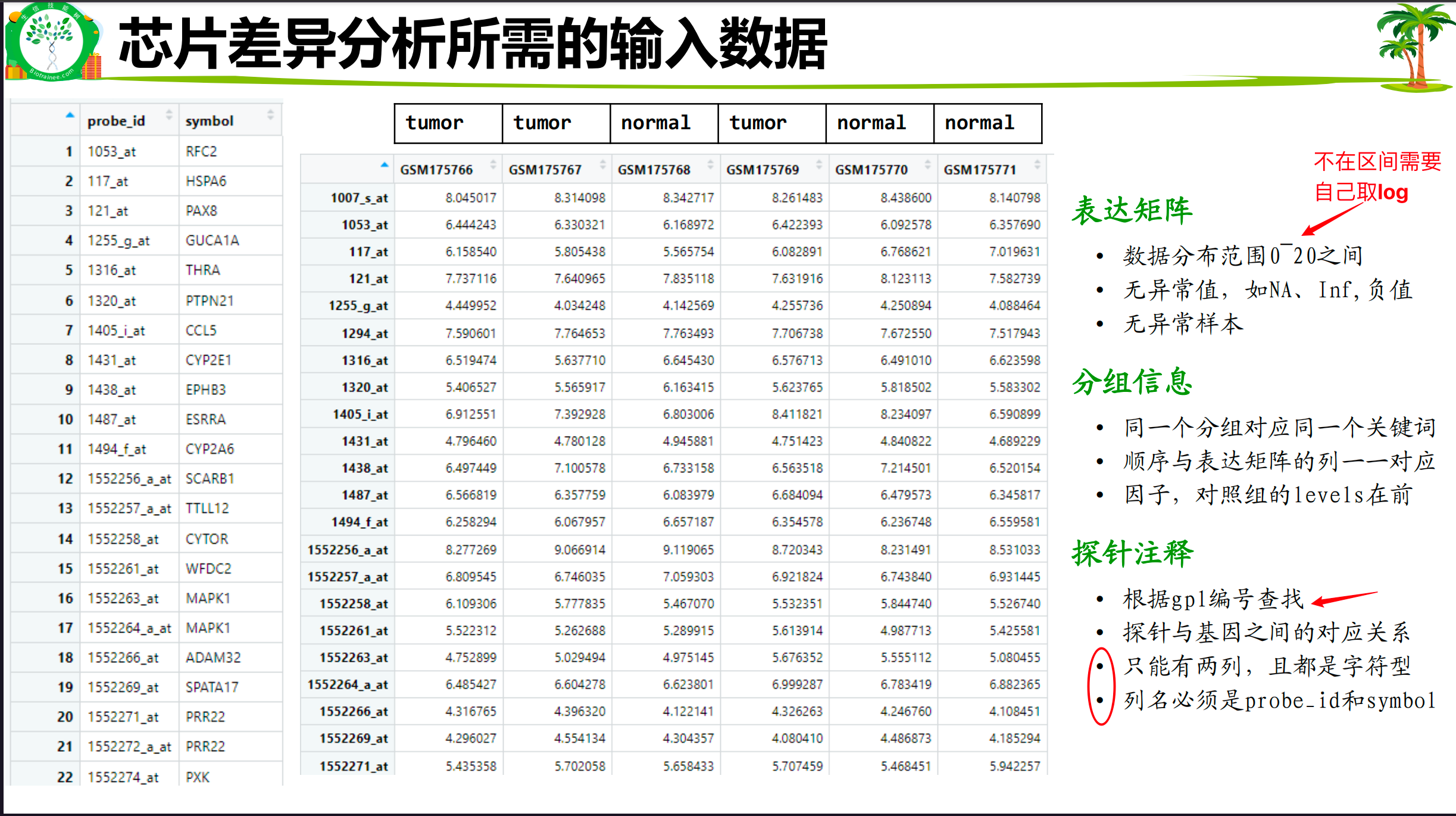
使用%in%可以检测探针注释是否对应
3.代码分析流程

分开代码好处,减少重复运行时间,降低检查结果的难度
##3.1 查找数据并提取数据信息
下载 Series Materix.txt并放在工作目录下
基因表达芯片的数据大小500k以下说明基因太少或者样本不正常
两种数据:常规转录组;单细胞/基因表达芯片
单细胞和常规转录组的数据在supplemental file里
3.1 具体代码01 数据下载并获取表达矩阵/芯片信息/分组信息
rm(list = ls())
#打破下载时间的限制,改前60秒,改后10w秒
options(timeout = 100000)
options(scipen = 20)#不要以科学计数法表示
#传统下载方式
library(GEOquery)
eSet = getGEO("GSE7305", destdir = '.', getGPL = F)#下载及/或读取文件
#网速太慢,下不下来怎么办
#1.从网页上下载/发链接让别人帮忙下,放在工作目录里
#2.试试geoChina,只能下载2019年前的表达芯片数据
#library(AnnoProbe)
#eSet = geoChina("GSE7305") #选择性代替第8行
#研究一下这个eSet
class(eSet)
length(eSet)
eSet = eSet[[1]] #使得列表的第一个元素重新赋值给eSet
class(eSet)
#(1)提取表达矩阵exp
exp <- exprs(eSet)#获取对象的表达矩阵
#⭐第一个要检查的地方👇,表达矩阵行列数,正常是几万行,列数=样本数,几百行比较难分析出东西
#如果0行说明不是表达芯片或者是遇到特殊情况,不能用此流程分析
dim(exp)#检查行列的函数
#⭐二个要检查的地方👇
range(exp)#看数据范围决定是否需要log,是否有负值,异常值,如有负值,结合箱线图进一步判断
#⭐可能要修改的地方👇
exp = log2(exp+1) #需要log才log,如果不需要log要注释掉这一句,exp+1是为了避免出现负值和0
#⭐第三个要检查的地方👇
boxplot(exp,las = 2) #看是否有异常样本
#(2)提取临床信息
pd <- pData(eSet)
#⭐多分组中提取两分组的代码示例,二分组不需要
if(F){
#因为现在这个例子不是多分组,所以编造一列做示例。
pd$fake = paste0(rep(c("a","b","c","d"),each = 5),1:5)
k1 = str_detect(pd$fake,"b");table(k1)
k2 = str_detect(pd$fake,"c");table(k2)
pd = pd[k1|k2,]
}
#(3)让exp列名与pd的行名顺序完全一致
p = identical(rownames(pd),colnames(exp));p
if(!p) {
s = intersect(rownames(pd),colnames(exp))
exp = exp[,s]
pd = pd[s,]
}
#(4)提取芯片平台编号,后面要根据它来找探针注释
gpl_number <- eSet@annotation;gpl_number
save(pd,exp,gpl_number,file = "step1output.Rdata")
# 原始数据处理的代码,按需学习 比较难
# https://mp.weixin.qq.com/s/0g8XkhXM3PndtPd-BUiVgw
下载基因芯片数据后修改为对象
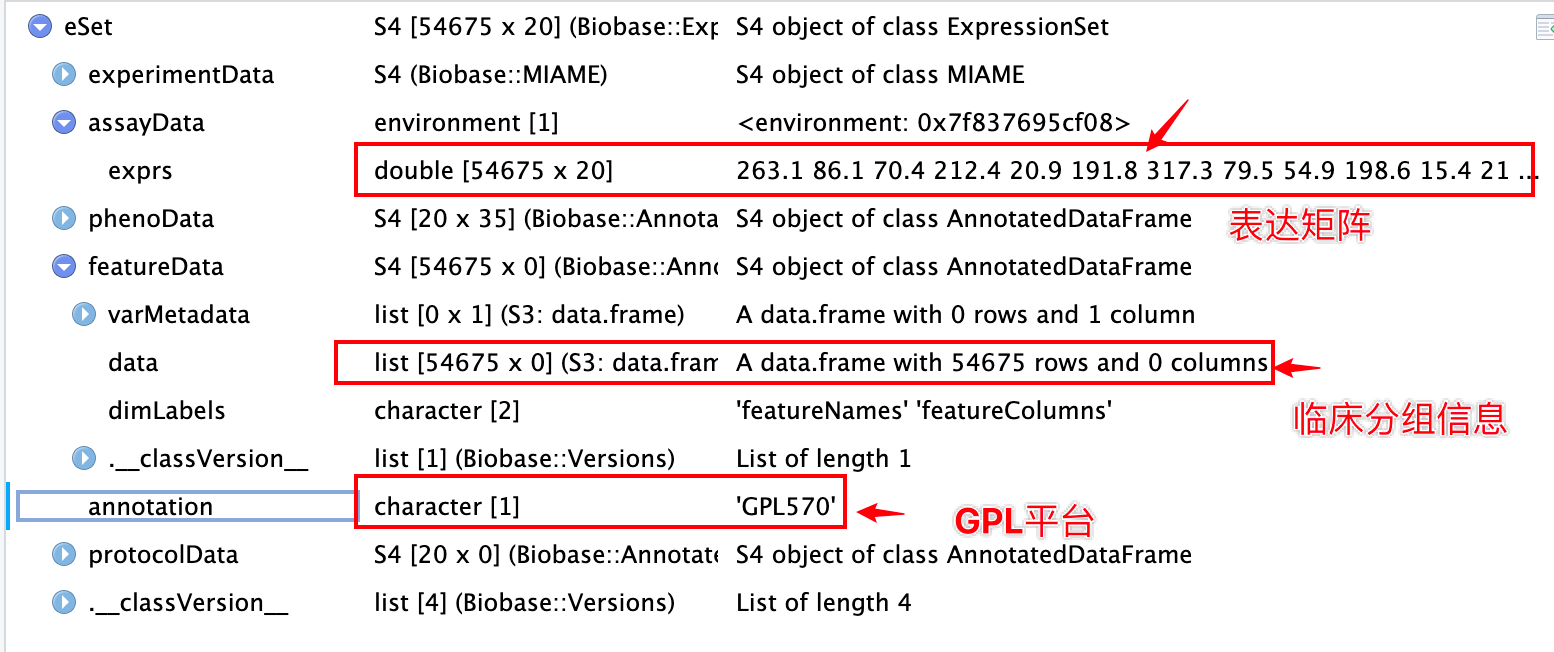
基因芯片对象里包含的内容
注意:修改代码后需要全选运行,避免漏运行,顺序错误,重复运行导致错误
箱线图判断原始数据:
1、钉子图提示表达量未取log2
2、异常样本:单个表达值均值明显低或高于其他样本
3、中位数在0附近,是不正常的zscore/scale标准划的数据且不能逆转(不能用于差异基因分析,但是可以做PCA/机器学习/热图)
4、没取过log且有负值:提示错误数据(建议换一个数据或处理原始数据)
5、取过1og,有少量的负数,但是4<中位数<15 这种数据正常
3、优先找靠谱正常数据 降低难度
4、代码需要修代的地方:修改GSE名;判断是否需要29行是否需要log
3.2 具体代码02 分组信息和探针注释
# Group(实验分组)和ids(探针注释)
rm(list = ls())
load(file = "step1output.Rdata")
# 1.Group----
library(stringr)
#⭐要修改的地方:分组信息,必须学会ifelse和str_detect
k = str_detect(pd$title,"Normal");table(k) #不在title就在pd的其他列
Group = ifelse(k,"Normal","Disease")
# 需要把Group转换成因子,并设置参考水平,指定levels(有利于后续差异基因分析明确对照组)
#⭐要修改的地方,对照组在前,处理组在后
Group = factor(Group,levels = c("Normal","Disease"))
Group
#⭐检查自己得到的分组是否正确
data.frame(pd$title,Group)
#2.探针注释的获取-----------------
#四种方法,方法1里找不到就从方法2找,以此类推。
#方法1 BioconductorR包(最常用)
#⭐要操作的地方
library(tinyarray)
gpl_number #首先看看编号是多少
#View(pkg_all)
#然后在pkg_all里搜索gpl编号,找到对应的R包前缀(第二列),没搜到就是没有R包,再看方法2。
#也可以用代码直接得到对应的R包前缀:
pkg_all[pkg_all$gpl==gpl_number,2]
#⭐要操作的地方
#用上面找到的前缀替换下面所有的hgu133plus2,共5处
if(!require(hgu133plus2.db))BiocManager::install("hgu133plus2.db",ask = F,update = F)
library(hgu133plus2.db)
ls("package:hgu133plus2.db") #列出R包里都有啥
ids <- toTable(hgu133plus2SYMBOL) #把R包里的注释表格变成数据框
# 方法2 下载并读取GPL网页的表格文件,按列取子集
#⭐要操作的地方
library(tinyarray)
get_gpl_txt(gpl_number) #获取表格文件的下载链接
# 接下来是复制网址去浏览器下载、放在工作目录下、读取、提取探针id和基因symbol(没有现成的需要拆分和转换),不同文件代码不统一,等看同学们的例子。
# 注意:最终的数据ids只能有两列,第一列列名是probe_id,第二列列名是symbol,且都是字符型,否则后面代码要报错咯。
# 方法3 官网下载注释文件并读取
# 方法4 自主注释,了解一下
#https://mp.weixin.qq.com/s/mrtjpN8yDKUdCSvSUuUwcA
save(exp,Group,ids,file = "step2output.Rdata")
#比较复杂的探针注释参考资料
#资料1:拆分取列https://www.yuque.com/xiaojiewanglezenmofenshen/kzgwzl/sv262capcgg9o8s5
#资料2:多种id的转换https://www.yuque.com/xiaojiewanglezenmofenshen/kzgwzl/pn0s1mmsaxocfynb?singleDoc# 《又一个有点难的探针注释(多种id的转换)》注意打开文件表格看,包含分组的列不一定是tittle
1/首先根据关键词定义分组
2/factor()将字符串转换为因子
3/确保对照组因子的levels在前面
4/找到探针注释对应的R包或者网站下载
怎么把上一步的数据保存成R.data:save(exp,Group,ids,file = "step2output.Rdata")
4、复杂数据分析流程
引用自用自生信技能树(小洁老师)
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号