RNA测序数据怎么定量?一篇给小白看的“白话”综述


把 RNA-seq 数据变成“每个基因/转录本有多少分子”这件事,目前主流有三条路:
- 1. 先比对再数 reads(alignment-based);
- 2. 不比对直接“扔骰子”估计分子数(pseudoalignment);
- 3. 用长读长技术直接数全长分子(long-read)。 下面按“原理—工具—注意事项—参考文献”的顺序聊,保证能看懂,能上手,能避坑。
一、为什么要“定量”而不是简单数 reads?
早期 RNA-seq 就是把 reads 比对到基因组,然后用 featureCounts 或 HTSeq-count 数落在每个基因上的 reads 数(Anders, 2015)。 问题:
- • 一条 read 可能同时匹配多个转录本(多重比对)。
- • GC 含量、基因长度、测序深度不同都会带来偏差(Dillies et al., 2013)。 因此,大家转而用概率模型把 reads 按概率分配到各转录本上,最后给出 TPM(每百万转录本数)或 expected count(期望计数),这才算“定量”。
二、短读长数据的两大门派
门派 | 核心思想 | 代表软件 | 特点(大白话) |
|---|---|---|---|
比对后定量 | 先 STAR/BWA 做比对 → 再数 reads | RSEM | 经典、稳,但硬盘和时间都要得多 |
Pseudo | 直接根据 k-mer 字典“猜” reads 来源 | Salmon , Kallisto | 快得飞起,几秒搞定一个样本,精度还挺好 |
1. Alignment-based 经典流程
Li & Dewey 2011 年提出的 RSEM 是目前最常用的“比对+期望最大化”框架(Li & Dewey, 2011):
- • 利用 Bowtie/STAR 把 reads 比对到“转录本参考序列”;
- • 用 EM 算法把多重比对 reads 按概率分配到各 isoform;
- • 输出 TPM + expected count。 优点:有成熟的下游差异分析生态(DESeq2、edgeR)。 缺点:比对步骤慢,硬盘大户。
2. Pseudoalignment 光速流派
Patro 等 2017 年的 Salmon 把“参考转录本”切成 k-mer 并建立索引,reads 过来后直接查字典,省掉比对(Patro et al., 2017)。
- • 速度提升 20–50 倍;
- • 内置 GC-bias、片段长度修正;
- • 输出同样是 TPM / NumReads。
Kallisto(Bray et al., 2016)思路类似,两者精度与 RSEM 几乎无差(Bray et al., 2016)。 小白上手建议:电脑一般就直接 Salmon / Kallisto,跑得快,硬盘省,命令行友好(Smith, 2019)。
三、长读长数据:直接数全长转录本
PacBio Iso-Seq 或 ONT 直接测出整条 mRNA,理论上不用再“猜”isoform。但实测发现:
- • 长读错误率高,需要校正;
- • 建库偏好仍在,需要定量模型(Jain et al., 2018)。
近两年出现了 IsoQuant(Prjibelski et al., 2023)和 Bambu(Chen et al., 2023)这类工具:
- • 先把长读比对到基因组,再用图模型或机器学习把 reads 聚成“可信转录本”;
- • 计算每个 isoform 的分子数,输出 TPM / read count(Prjibelski et al., 2023)。
系统评估显示,长读长定量在复杂基因位点(>10 isoforms)时比短读更准(Chen et al., 2023)。
四、结果怎么看?单位科普
指标 | 含义(一句话) | 用途 |
|---|---|---|
raw counts | 落在每个基因的 reads 数,未校正 | 不能直接比 |
TPM | 每百万转录本数,已校正基因长度和测序深度 | 同一基因在不同样本比 |
FPKM/RPKM | 早期单位,现在基本被 TPM 取代 | 别用了,会踩坑 |
差异分析工具(DESeq2、edgeR)只吃 raw counts,它们自己会做归一化(Love et al., 2014)。
五、常见坑位提醒
- 1. 参考基因组/转录本版本要一致 GENCODE v39 vs vM28 混用会导致定量天差地别(Frankish et al., 2021)。
- 2. 重复序列多的基因(例如 SNORD 簇) 短读几乎无法区分,长读或靶向捕获更靠谱(Zhang et al., 2020)。
- 3. 单端 vs 双端 Salmon/Kallisto 都能吃单端,但差异分析时记得在 colData 里标注(Smith, 2019)。
- 4. ERCC spike-in 如果实验加了外部 RNA 标准品,可用它来评估定量线性范围(Jiang et al., 2011)。
六、思维导图总结
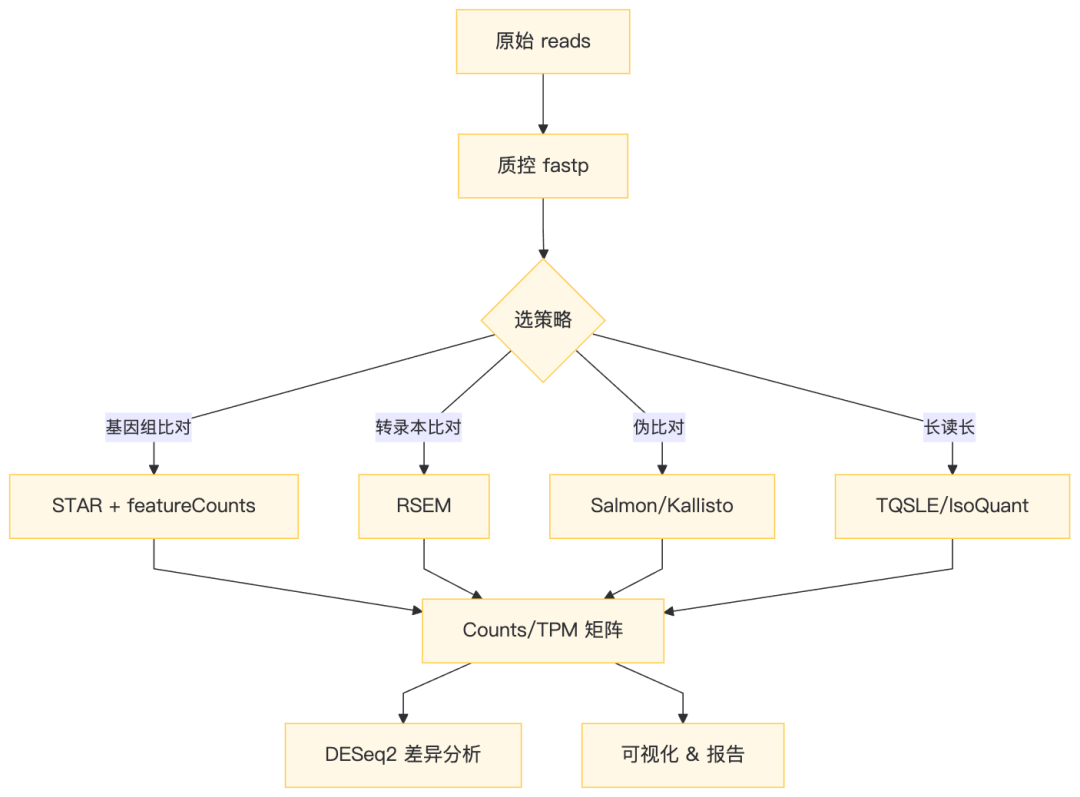
七、参考文献
- • Anders, S. (2015). HTSeq documentation: counting reads in features. HTSeq GitHub.
- • Bray, N. L. et al. (2016). Near-optimal probabilistic RNA-seq quantification. Nature Biotechnology, 34, 525–527.
- • Chen, Y. et al. (2023). Context-aware transcript quantification from long-read RNA-seq data with Bambu. Nature Methods.
- • Dillies, M. A. et al. (2013). A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Briefings in Bioinformatics, 14, 671–683.
- • Frankish, A. et al. (2021). GENCODE 2021. Nucleic Acids Research, 49(D1), D916–D923.
- • Jain, M. et al. (2018). Nanopore sequencing and assembly of a human genome with ultra-long reads. Nature Biotechnology, 36, 338–345.
- • Jiang, L. et al. (2011). Synthetic spike-in standards for RNA-seq experiments. Genome Research, 21, 1543–1551.
- • Li, B. & Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics, 12, 323.
- • Love, M. I. et al. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology, 15, 550.
- • Patro, R. et al. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nature Methods, 14, 417–419.
- • Prjibelski, A. D. et al. (2023). Accurate isoform discovery with IsoQuant using long reads. Nature Biotechnology.
- • Smith, T. (2019). Salmon vs. Kallisto: a quick guide for beginners. Biostars Blog.
- • Zhang, Y. et al. (2020). Model-based analysis of ChIP-Seq (MACS). Genome Biology, 9, R137.
社区简介
中国银河生信云平台(UseGalaxy.cn)以“让生信分析更简单”为使命。平台致力于为科研工作者、医疗机构和生物产业技术人员提供全栈式生物信息学分析解决方案。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-08-03,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号