巨型病毒识别工具——GiantHunter教程


Hello,Hello小伙伴们大家好,好久没有给大家更新微信公众文了,最近小编在搭建宏病毒组相关流程,宏病毒和宏基因最大的区别在于,在组装的fasta序列中识别病毒序列。今天小编要给大家介绍一款从宏基因组中识别巨病毒序列的工具——GiantHunter,这款工具由香港城市大学孙燕妮课题组的博士生商家煜开发,专为解决宏基因组中巨病毒识别难题而设计,非常值得推荐!
巨型病毒属于双链DNA病毒的一部分,被统称为核质大DNA病毒(Nucleocytoplasmic Large DNA Viruses, NCLDVs),系统分类上归入病毒门 Nucleocytoviricota。该门病毒具有高度的宿主多样性,可感染从最小的单细胞真核生物(如领鞭毛虫)到多细胞动物在内的多种真核宿主。NCLDVs 的复制通常在宿主细胞质中构建的“病毒工厂”中进行,部分成员也可利用宿主细胞核进行复制和子代组装。
NCLDVs 的一个显著特征是其粒子尺寸和基因组大小的高度扩展性:基因组范围从约 70 kb 到超过 2.5 Mb 不等,病毒粒子长度可达 2 μm,是目前所知最大尺寸的病毒粒子之一。术语“巨型病毒”(Giant Virus)最早可追溯至20世纪90年代,用于描述感染藻类、具有异常大基因组的病毒。
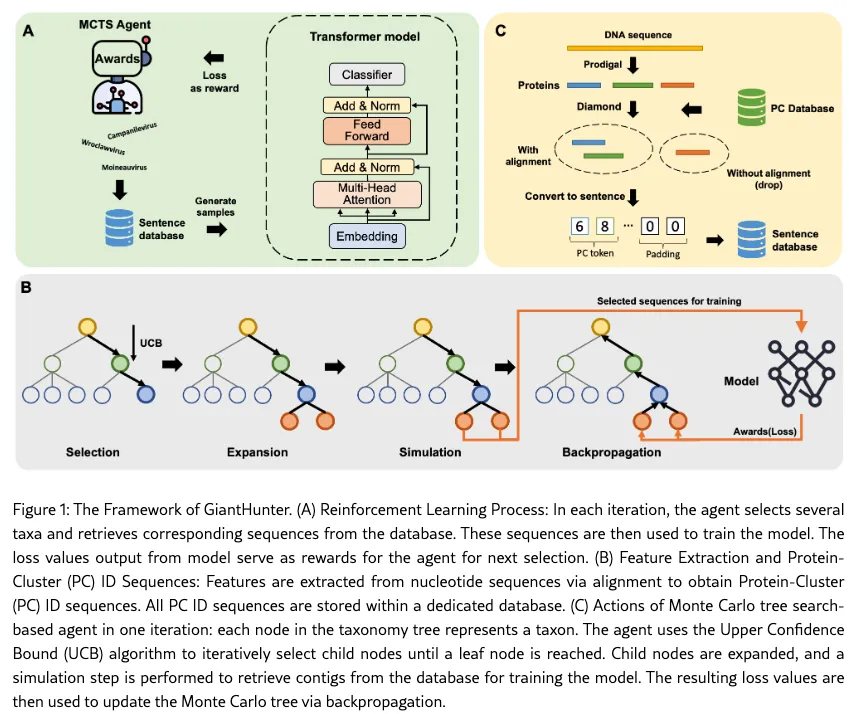
软件原理
Github:
https://github.com/FuchuanQu/GiantHunter
数据库:
https://github.com/FuchuanQu/GiantHunter/releases/download/v2.0/gianthunter_db_v1.zip
软件安装
# 从Github下载源码
git clone https://github.com/FuchuanQu/GiantHunter.git
# 进入目录
cd GiantHunter/
# 使用conda从创建虚拟环境
conda env create -f GiantHunter.yaml -n GiantHunter
# 激活环境
conda activate GiantHunter
# 安装GiantHunter,GiantHunter本质是一个python模块,可以使用pip安装
pip install .
# 检查是否安装成功
gianthunter -h
# 下载数据库并解压
wget -c -nv https://github.com/FuchuanQu/GiantHunter/releases/download/v2.0/gianthunter_db_v1.zip
unzip gianthunter_db_v1.zip注:
1.小编的安装方法和作者在Github上提及的方法有所不同,因为小编习惯将每个软件单独创建一个虚拟conda环境。作者在Github上提及到的方法将GiantHunter和PhaBOX 2安装在一起,因为这两个软件都是一个团队开发的,两款软件依赖的软件相同;
2.如果小伙伴们在运行GiantHunter中缺少相关的python模块,可使用pip进行安装;
3.如果数据下载太慢,小伙伴们可以使用浏览器下载数据库,然后传到服务器上,此外一些小伙伴无法科学上网,作者也将数据库传到百度网盘上,小伙伴也可以通过百度网盘进行下载,百度网盘下载如下:
https://pan.baidu.com/s/1YJBaXA0OmvUfXEK8QEnkDg
pwd:jrif软件使用方法
# 查看软件帮助
gianthunter -h
usage: gianthunter [-h] [--contigs CONTIGS] [--proteins PROTEINS] [--len LEN] [--threads THREADS] [-d DBDIR] [--midfolder MIDFOLDER] [-o OUTPTH] [--reject REJECT] [--query_cover QUERY_COVER]
GiantHunter is a python library for identifying NCLDVs from metagenomic data. GiantHunter is based on a Transorfer model and relies on protein-based vocabulary to convert DNA sequences into sentences.
optional arguments:
-h, --help show this help message and exit
--contigs CONTIGS FASTA file of contigs
--proteins PROTEINS FASTA file of predicted proteins (optional)
--len LEN minimum length of contigs
--threads THREADS number of threads to use
-d DBDIR, --dbdir DBDIR
database directory (optional)
--midfolder MIDFOLDER
folder to store the intermediate files
-o OUTPTH, --outpth OUTPTH
name of the output folder
--reject REJECT threshold to reject contigs with a small fraction of proteins aligned.
--query_cover QUERY_COVER
The QC value set for DIAMOND BLASTP, setting to 0 means no query-cover constrain.常用参数解读:
参数 | 说明 |
|---|---|
--contigs CONTIGS | 输入的 contig 文件,格式为 FASTA |
--proteins PROTEINS | 可选,预测得到的蛋白质序列(FASTA 格式),加快软件运行时间 |
--len LEN | 可选,contig 的最小长度阈值,低于该长度的 contig 将被过滤,默认为3000 |
--threads THREADS | 可选,使用的线程数,可以使用 os.cpu_count() 设置为最大核数 |
-d DBDIR, --dbdir DBDIR | 数据库路径 |
--midfolder MIDFOLDER | 可选,中间文件的存储路径,有助于调试或重复使用中间结果 |
-o OUTPTH, --outpth OUTPTH | 指定输出结果路径 |
--reject REJECT | 可选,设置一个阈值,如果某 contig 上比对成功的蛋白占比低于此值,则剔除,默认为0.1 |
--query_cover QUERY_COVER | 可选,用于 DIAMOND BLASTP 的 query-cover 设置,默认为40 |
实战演练
gianthunter --contigs NCBI.Virus.fna --len 2000 --threads 16 --query_cover 40 --dbdir db --out GiantHunter示例数据来源:小编在NCBI virus:https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/随机选取一些巨型病毒和噬菌体序列作为模拟数据,模拟数据获取方法,关注公众号,后台回复后台回复关键字【GiantHunter】 (不含中括号哟),建议粘贴复制,避免出错。
结果文件说明
1.结果目录
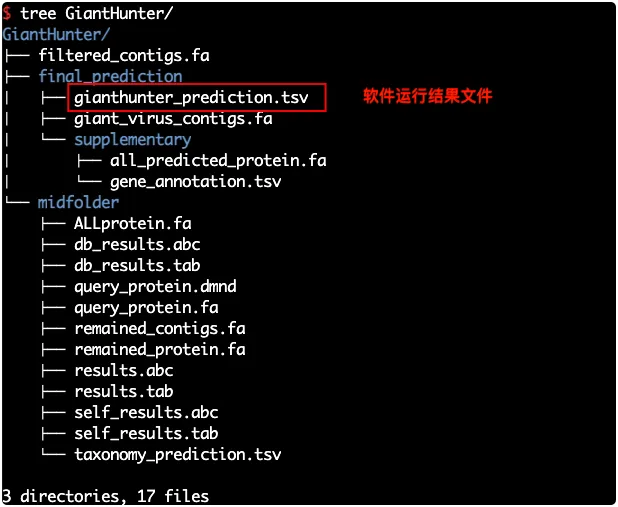
结果解读:
gianthunter_prediction.tsv————记录了每个 contig 的预测得分和判定结果- all_predicted_protein.fa——使用prodigal-gv预测出来的蛋白质序列
- gene_annotation.tsv——蛋白质功能注释结果
2. gianthunter_prediction文件内容如下图所示
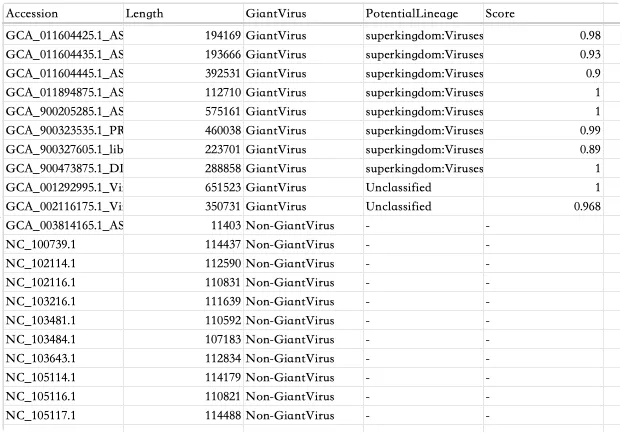
- Accession:Contig编号;
- Length:Contig长度(碱基数);
- GiantVirus:是否属于巨病毒,"GiantVirus"或"Non-GiantVirus";
- PotentialLineage:巨病毒物种注释信息;
- Score:置信度评分或分类可信度,范围 0-1。
参考文献
- La Scola B, Audic S, Robert C. et el. A giant virus in amoebae. Science. 2003 Mar 28;299(5615):2033. doi: 10.1126/science.1081867.
- Philippe N, Legendre M, Doutre G .et el. Pandoraviruses: amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science. 2013 Jul 19;341(6143):281-6. doi: 10.1126/science.1239181. Erratum in: Science. 2013 Sep 27;341(6153):1452.
- Schulz F, Abergel C, Woyke T. Giant virus biology and diversity in the era of genome-resolved metagenomics. Nat Rev Microbiol. 2022 Dec;20(12):721-736. doi: 10.1038/s41579-022-00754-5. Epub 2022 Jul 28.
- Qu F, Peng C, Guan J, Wang D, Sun Y, Shang J. GiantHunter: Accurate detection of giant virus in metagenomic data using reinforcement-learning and Monte Carlo tree search. arXiv preprint arXiv:2501.15472. 2025 Jan 26.
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-06-27,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号