提供代码数据,向Nature学习如何用基因组数据来分析癌症的演化轨迹

提供代码数据,向Nature学习如何用基因组数据来分析癌症的演化轨迹

生信菜鸟团
发布于 2025-05-09 10:35:48
发布于 2025-05-09 10:35:48

Basic Information
- 英文标题:Timing and trajectory of BCR::ABL1-driven chronic myeloid leukaemia
- 中文标题:BCR::ABL1驱动的慢性髓性白血病的发生时间和轨迹
- 发表日期:09 April 2025
- 文章类型:Article
- 所属期刊:Nature
- 文章作者:Aleksandra E. Kamizela | Jyoti Nangalia
- 文章链接:https://www.nature.com/articles/s41586-025-08817-2
Abstract
某些基因的突变会驱动细胞不受控制地增殖并导致癌症。慢性髓性白血病(CML)中的费城染色体首次揭示了这种基因与癌症的关联<sup>1,2</sup>。然而,关于CML的发病轨迹、BCR-ABL1融合基因克隆扩增的速率及其如何影响疾病的认知仍然有限。通过对9名年龄在22至81岁之间的CML患者的1013个造血集落进行全基因组测序,我们重建了造血过程的系统发育树。研究发现,BCR和ABL1基因的内含子断裂并非总是出现,同时也观察到BCR基因中存在框外外显子断裂点,这需要通过外显子跳跃来形成BCR-ABL1融合基因。除了ASXL1和RUNX1突变外,其他额外的髓系基因突变主要存在于野生型细胞中。我们推断,由BCR-ABL1引起的爆发性增长在诊断前3-14年(置信区间为2-16年)即已开始,年增长率超过70,000%。在端粒长度较短的BCR-ABL1细胞中,突变积累更高,这反映了其过度的细胞分裂。克隆扩增速率与诊断时间呈负相关。普通人群中BCR-ABL1的检出情况与CML的发病率相符,而晚期和/或急变期CML的特征在于后续的基因组进化。这些数据突显了BCR-ABL1融合基因的强大致癌潜能,并与大多数癌症缓慢且连续的克隆演化轨迹形成了对比。
Main
Para_01
- 慢性髓性白血病(CML)在肿瘤学研究史上占据了一个里程碑的位置,标志着首次发现遗传异常与癌症的发展有关。
- 1960年,Nowell和Hungerford1发现了费城(Ph)染色体,1973年,Rowley2发现了BCR::ABL1融合基因,这预示了肿瘤基因组学时代的到来。
- 通过酪氨酸激酶抑制(TKI)靶向BCR::ABL1,自那时起在CML患者中取得了独特且成功的治疗效果,这一成果尚未被大多数其他癌症所复制。
Para_02
- 癌症是由关键基因突变的逐步积累导致的,这些突变对细胞生长和调控至关重要。
- 这种突变会在很长一段时间内累积,在临床表现之前的几十年就开始了,例如,卵巢癌早期会出现全基因组复制,而透明细胞肾细胞癌则会出现3号染色体3p的丢失。
- 癌症的进化甚至可能始于子宫内,这一点在成人发作的多血症患者中JAK2突变的研究中得到了证明。
- 相比之下,日本原子弹幸存者的癌症发病率显示,在辐射暴露后的10年内慢性髓性白血病(CML)达到了峰值,这引发了BCR::ABL1驱动的克隆扩增和向CML发展的轨迹与迄今为止研究的成人恶性肿瘤不同的可能性。
Para_03
- 体细胞突变在整个生命过程中以类似时钟的方式在造血细胞中积累。
- 由此产生的单个细胞的独特突变谱可以用来重建描绘祖先细胞关系和进化历史的系统发生树。
- 这种方法已经能够精确量化健康造血和血液恶性肿瘤中的克隆动力学。
- 在这里,我们利用全基因组体细胞突变和系统发生推断,描述了BCR::ABL1驱动的克隆扩展的适应度和轨迹,以及这些因素如何影响CML的临床特征。
Driver mutations in CML colony genomes
Para_01
- 我们研究了九名患者,年龄在初诊时为22至81岁,患有慢性期CML(图1a和补充表1)。
- 患者携带BCR::ABL1易位t(9;22)(q34;q11),具有典型的融合转录物e14a2或e13a2,涉及BCR的主要断裂簇区。
- 患者对TKI治疗的反应各不相同。
- 三名患者对一线伊马替尼或达沙替尼治疗有响应,六名患者需要二线TKI治疗,两名患者未能通过多种不同的TKI治疗。
- 在九名患者中的六名中,在诊断时采集了血液和骨髓样本,并且在四名患者中进一步在治疗期间进行了采集(图1a)。
- 在三名个体(PD57333、PD57334和PD57335)中,采样时间点仅在诊断后(7个月到2年9个月,图1a和补充表1)。
- 单核细胞(MNCs)体外培养以提供单细胞衍生克隆DNA用于全基因组测序(WGS)(图1b)。
- 经过质量控制,由于测序覆盖度低或克隆性降低,1013个菌落中有179个被排除。
- 总共834个全基因组被纳入分析(每名患者平均93个全基因组,范围35-163,测序深度平均15.3倍,范围8.6-46.3倍)。
- 共鉴定出397,063个常染色体单核苷酸变异(SNVs)和13,646个常染色体插入和缺失。
Fig. 1: Patient cohort and study design.
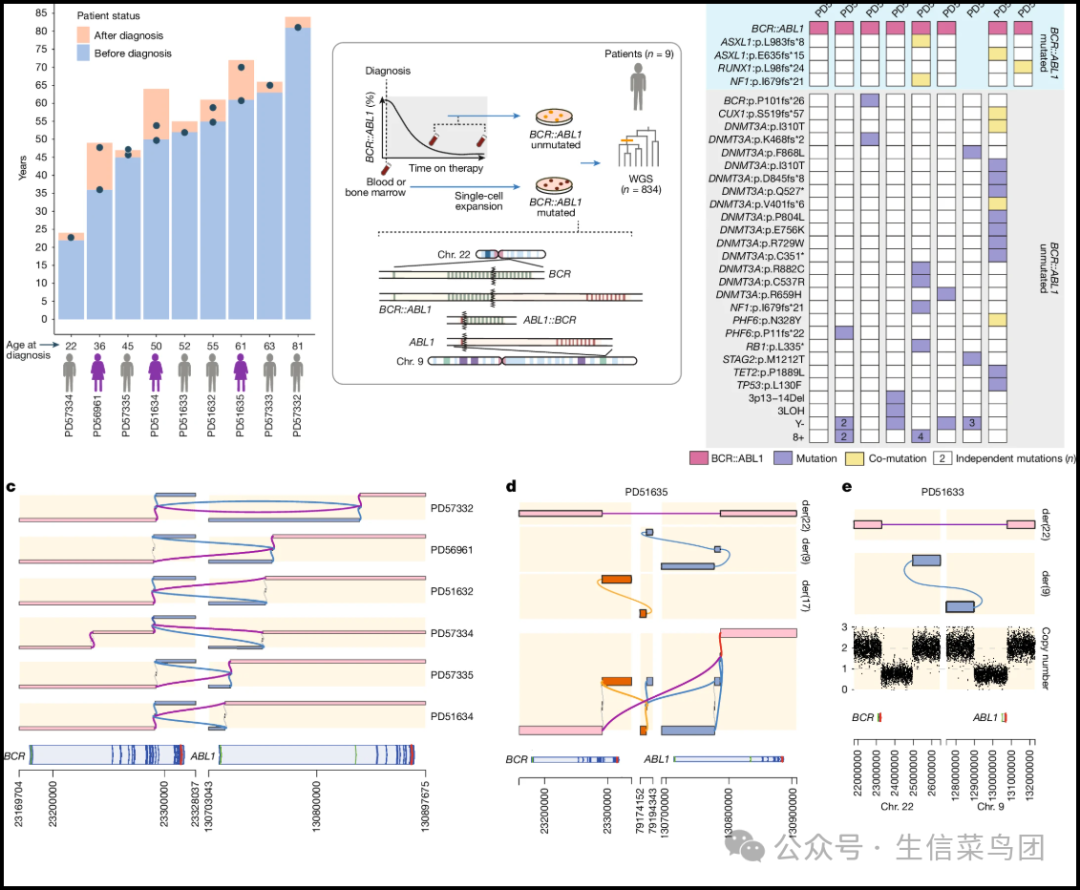
- 图片说明
◉ 患者队列。条形长度对应患者的最后随访或死亡年龄,橙色阴影表示疾病持续时间,蓝色阴影表示诊断前的时间。圆点代表取样时间点。◉ 实验设计,展示了t(9;22)易位,导致BCR::ABL1融合(费城染色体或衍生染色体22,der(22))和反向的ABL1::BCR融合(der(9))的示意图。◉ 典型的、相互的BCR::ABL1结构变异图,在八名患者中的六名中存在。der(22)用粉红色表示,并用紫色路径从染色体9到染色体22,而der(9)用蓝色表示,并用蓝色路径从染色体22到染色体9。◉ 任何没有紫色或蓝色路径的部分表示参考基因组的丢失:这种小的丢失(1-49 bp)发生在六名患者中的五名。◉ 任何既有紫色又有蓝色路径的部分表示参考基因组的重复。◉ 患者PD51635中的BCR::ABL1涉及染色体17,产生非典型der(9),用蓝色表示,der(17)用橙色表示,der(22)用粉红色表示。◉ PD51633中的BCR::ABL1涉及der(9)上的大缺失,排除了ABL1::BCR的形成。下图显示了染色体22和染色体9上大片段的拷贝数从两个减少到一个,表示被删除的区域。◉ 每个患者检测到的至少在一个克隆中存在的驱动突变和拷贝数改变。粉色方块表示识别出的BCR::ABL1易位,紫色方块表示患者内的单一突变,黄色方块表示该突变发生在携带不同突变的克隆内。◉ 在CML克隆内观察到的突变显示在顶部的蓝色阴影区域内,而在野生型克隆内存在的突变显示在底部的灰色阴影区域内。◉ 数字代表患者内给定基因独立获得的突变数量。
Para_02
- 我们在九个人中的八个人中检测到了BCR::ABL1(图1c-e)。
- 在PD57333中,在分子上可检测到的复发时(BCR::ABL1/ABL1比率为3%),我们仅在全血WGS中捕获了BCR::ABL1(平均测序深度45.9倍下的两个DNA读对),但未从培养的菌落中捕获,因为突变克隆比例较低。
- 由于能够重建精确的基因组BCR::ABL1(衍生型,der(22))和ABL1::BCR(der(9))断裂点,WGS提供了独特的机会来调查BCR::ABL1断裂点,而常规临床互补DNA(cDNA)分析无法获得这些断裂点。
- 有两个观察结果明显。
- 首先,在融合点处观察到删除或复制的DNA区域(长度为1至687个碱基对(bp)跨越der(22)和der(9))。
- 在两名患者中,这完全破坏了der(9)。
- 在PD51635中,存在一个复杂的三向易位,导致形成der(22),但也导致t(9;17)(q34.12;q25.3)和t(17;22)(q25.3;q11.23),从而形成一个非典型的der(9)(图1d和补充表2)。
- 在PD51633中,我们观察到大约1Mb的大规模丢失,同时删除了5' ABL1和3' BCR以及55个额外基因(图1e和补充表2)。
- 我们还观察到在融合点处有非模板插入序列(扩展数据图2b、c)。
- 其次,虽然有几个个体在BCR和ABL1内含子中携带了预期的断裂点,但我们观察到三例(PD56961、PD57332和PD57335)在BCR中有外显子断裂点,这种情况在文献中很少报道。
- 所有三例都预测出了框外融合与ABL1结合,并且如果继续融合读码框将导致在1到17个密码子内出现终止密码子(扩展数据图2a-c)。
- 尽管如此,这些患者仍具有典型且可临床检测到的融合转录物(扩展数据图2d)。
- 这些融合转录物只有在框外外显子被剪接掉的情况下才能与来自DNA的事件兼容。
- 使用SpliceAI,所有病例显示框外外显子的剪接受体概率降低(PD56961的Δ分数为0.49,PD57332的Δ分数为0.67,PD57335的Δ分数为0.75),支持这一假设(扩展数据图2e)。
- 补充表2:
Para_03
- 在BCR::ABL1阳性的菌落中,我们在PD51635和PD51634中偶尔发现了ASXL1中的进一步突变(L983fs8,E635fs15)。
- 在PD57332中,在获得BCR::ABL1后,一个典型的RUNX1 RUNT结构域截断突变作为主要亚克隆重现(图1f和图2)。
- 据报道,这些驱动突变会影响患者的预后、治疗反应和疾病转化13,14,15,16。
- 进一步的驱动突变更常见于BCR::ABL1阴性的菌落中,尤其是在三体8(六次事件)、DNMT3A突变(十四次事件)和Y染色体丢失方面,但也包括TET2、NF1、CUX1、STAG2、PHF6、BCR、RB1和TP53中的突变(图1f和图2)。
- 或许,这些基因中的突变不一致地与CML特征和结果相关联并不令人惊讶15,16,17,因为它们并不是优先在CML克隆中被观察到的。
Fig. 2: Phylogenetic trees of chronic phase CML.
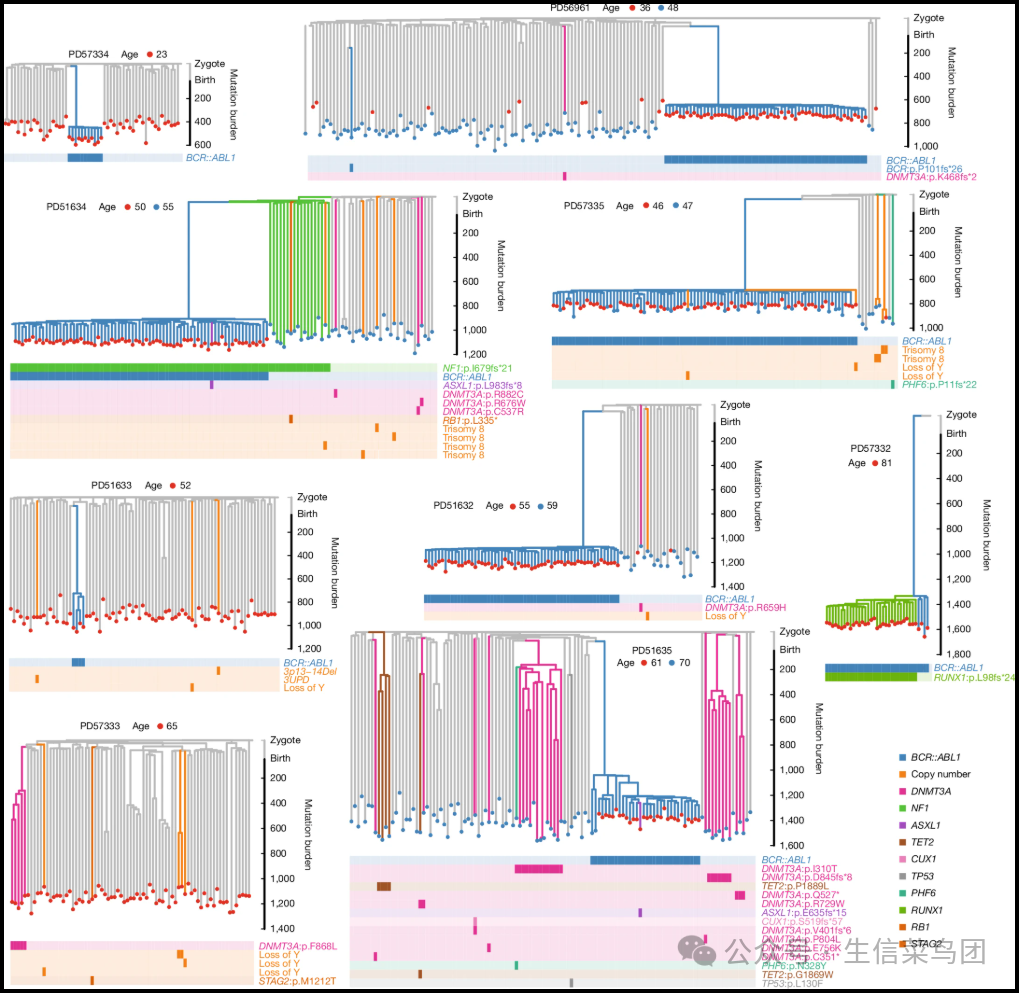
- 图片说明
◉ 每棵树展示了在采样菌落中体细胞突变的共享模式。◉ 右侧的纵轴显示了树中任意一点的体细胞突变负荷。◉ 私有分支代表仅存在于单一菌落中的突变,而共有分支代表存在于一个或多个菌落中的突变。◉ 树中的分叉点(共线)代表了一个祖先的造血干细胞,该细胞进行了一次对称的自我更新分裂,其中每个子细胞的后代被采样为一个菌落,并代表下游菌落或多克隆中的最近共同祖先(MRCA),或共享此MRCA的‘克隆群’。◉ 底部的圆点代表单个测序的菌落,它们的颜色对应于采样时间点。◉ 突出显示的分支由其携带的驱动突变和拷贝数变化着色。◉ 数据来源
Phylogenetic trees in patients with CML
Para_01
- 费城染色体阳性慢性髓性白血病(CML)九名患者的造血细胞系统发育树如图2所示,展示了整个基因组体细胞突变在各个单个群体中的共享模式。
- 共享的分支代表在下游子代谱系中发现的突变,而最低分支中的突变仅出现在单一群体中。
- 可以做出几个一般性观察。
- 正如预期的那样,个体群体中的突变总数随着患者年龄的增长而增加,这与人类造血干细胞和祖细胞(HSPCs)的类似时钟样的突变获取一致9,10,18。
- 所有的系统发育树在其根部或‘顶部’附近都有大量的合并点(分支点)。
- 这反映了造血干细胞的快速扩张以及胚胎发生过程中产生的基因组多样性,使得任何两个正常的造血干细胞,它们最近的共同祖先(MRCA)接近生命的开始。
- 在早期扩张之后,年轻患者的正常造血具有类似于‘梳子’的系统发育结构,而年长患者则有许多克隆扩张9。
- 我们观察到特定驱动突变的显著患者内重复出现,例如,在PD51635中有八个独立的DNMT3A突变,在PD57333中有三个独立的Y染色体丢失事件,在PD51633中有两个独立的3号染色体异常,在PD51634中有反复的8号三体现象,强烈表明存在患者特有的选择景观。
Para_02
- 这些树木的一个显著特征是BCR::ABL1阳性菌落的分支模式。
- 这些菌落在一条长而垂直的树枝下方出现,这条树枝只携带了所有分配到该树枝的所有体细胞突变中的一种已识别驱动因素:BCR::ABL1易位。
- 在六名患者中,BCR::ABL1阳性分支有一个快速合并的爆发,直接出现在这个共享分支之下,类似于生命开始时观察到的情况,表明迅速分裂成大量白血病HSPCs。
- 在两名患者(PD51635和PD51633)中,观察到相对较慢但仍然最近的克隆扩展模式。
- 这些数据显示,在慢性髓性白血病患者中,最近共同祖先(MRCA)和肿瘤克隆扩展的开始是近期事件,这与在造血过程中观察到的其他驱动突变的模式形成对比。
Mutation accumulation in BCR::ABL1 cells
BCR-ABL1阴性造血干/祖细胞(HSPCs)以每年18个突变(95%自举置信区间,CI<sub>boot</sub>,16.1–20.2)的速率累积突变,这与已发表的数据<sup>7,9</sup>一致。BCR-ABL1阳性HSPCs则额外多出约90个突变(平均+91.7,CI<sub>boot</sub> 33.6–151.6,图3a和补充说明1)。事实上,在许多患者中,BCR-ABL1阳性集落的突变负荷与BCR-ABL1阴性HSPCs相当甚至更高,尽管这些阳性集落被采样时其(克隆)年龄更小(例如患者PD51634和PD51632的细胞)(图2和图3a)。
Fig. 3: Mutation accumulation in BCR::ABL1 cells.
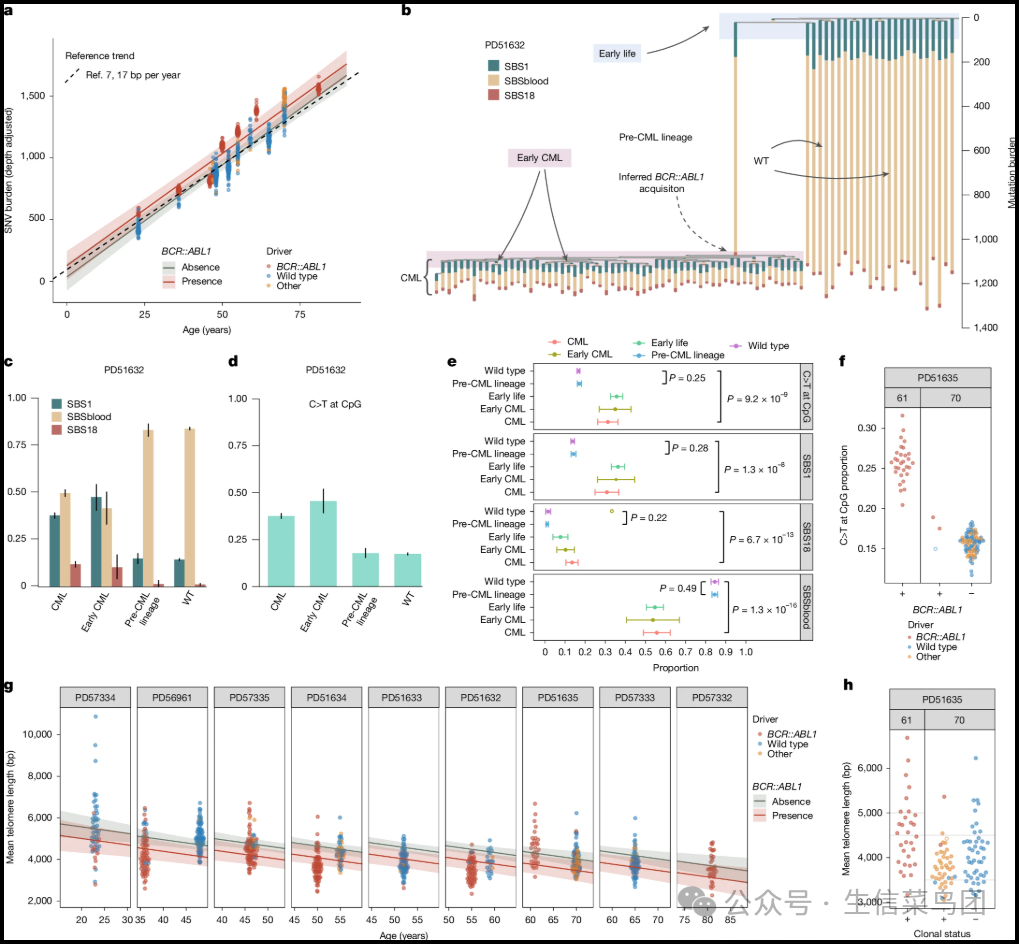
- 图片说明
◉ a, 来自菌落的体细胞SNVs数量(n = 834),根据驱动状态(BCR::ABL1,红色;其他驱动突变,橙色;野生型(WT),蓝色)和与年龄的关系进行着色,并通过混合效应模型回归线和按BCR::ABL1状态分层的95%置信区间表示(红色,BCR::ABL1阳性;灰色,BCR::ABL1阴性,由于缺乏野生型,模型排除了PD57332,n = 799)。◉ b, 一个患者的系统发育树示例PD51632,显示了每个分支上SBS签名SBS1、SBSblood和SBS18对SNVs贡献的估计比例,分为五组:(1)BCR::ABL1阳性克隆扩展('CML')的分支,(2)代表CML起源谱系的BCR::ABL1菌落祖先分支('前CML谱系'),(3)CML内的早期突变(即,在克隆扩展内的共享分支,'早期CML',下紫色阴影框),(4)BCR::ABL1阴性菌落(野生型,WT)的分支,(5)早期生活中的突变,代表系统发育树上的前100个突变,上蓝色阴影框。◉ c, PD51632的SBS1、SBSblood和SBS18对SNV光谱贡献比例的条形图,条形的高度表示后验均值,误差棒表示95%可信区间。◉ d, PD51632中C>T在CpG变化中的比例条形图。条形的高度表示观察到的比例,误差棒表示95%二项比例置信区间。◉ e, 排除PD57333和PD57332的队列范围内的C>T在CpGs、SBS1、SBS18和SBSblood比例图,因为这些患者缺乏野生型或突变菌落。点表示混合效应元分析估计的比例,条形表示95%置信区间。P值是对零假设的双侧检验,该假设认为比例之间的差异为零。P值未针对多重测试进行调整。◉ f, PD51635的每个样本点图,显示了C>T在CpG比例(排除BCR::ABL1阳性菌落中的BCR::ABL1分支克隆主干)的样本BCR::ABL1状态,按采样时间分面并注释了每个样本的驱动状态。◉ g, PD51635中每个样本的平均端粒长度(bp)按采样时的年龄分类,标注了驱动状态和按BCR::ABL1状态分层的混合效应模型回归线及95%置信区间。◉ h, PD51635中描述平均端粒长度(bp)按克隆状态(BCR::ABL1,另一种驱动突变或'无驱动'克隆扩展)的每个样本点图,按采样时间分面并注释了每个样本的驱动状态。
Para_02
- 我们认为这些增加的突变是由于新的或现有的突变过程引起的。
- 在BCR::ABL1阳性和阴性的菌落中,除了SBS18之外,还存在预期的内源性时钟样突变过程(单碱基替换(SBS)特征SBS1和SBSblood)(图3b、c)。
- 没有发现额外的突变过程,这与之前关于BCR::ABL1诱导的DNA损伤的报告相反(20)。
- 然而,我们没有发现TKI诱导的突变,但在BCR::ABL1阴性的菌落中观察到了反复出现的8号染色体三体性(图1f和2)。
- 这可能反映了在CML发展过程中8号染色体造血干细胞的增殖或生存增加,或者随后被TKI选择性地正向选择。
Para_03
- 为了探索现有的突变过程如何导致BCR::ABL1阳性细胞中的突变负担增加,我们将突变特征分配给系统发育树的各个分支(图3b和扩展数据图3和4),按(1)BCR::ABL1阴性(野生型)分支,(2)代表前CML系的BCR::ABL1克隆共享分支,(3)代表早期CML克隆扩增的历史早期BCR::ABL1克隆共享分支,(4)包括最近突变的所有BCR::ABL1克隆分支,以及(5)生命早期分支。
- 在BCR::ABL1克隆中观察到SBS1突变的比例增加(图3c和扩展数据图3,+13.6–30.9%,P=4.0×10^-11)。
- 这与在CpG位点观察到的C>T转换比例增加相呼应(图3d和扩展数据图4,+16.6–31.3%,P=2.9×10^-11)。
- 这反映了由于细胞分裂增加导致的甲基化胞嘧啶自发脱氨基作用增加(图3e)。
- 类似的现象也出现在SBS18中,该现象在胎儿造血快速生长环境中19和胎盘组织22中也有观察到。
- 总体而言,我们的数据显示,CML细胞中的突变增加仅仅是反映了它们更高的细胞分裂率。
Para_04
- 我们观察到,在前CML谱系(BCR::ABL1分支的共享分支)和野生型HSPCs之间没有发现突变模式的差异(图3e)。
- 这证实了CML起源细胞在被BCR::ABL1转化之前获得了与正常HSPCs相似的突变,这可能发生在长共享分支的末端。
Para_05
- 多年后诊断出BCR::ABL1阳性的菌落是不寻常的,因为大多数患者处于分子缓解状态。
- PD51635具有临床稳定的低BCR::ABL1/ABL1比率(0.5%)在TKI治疗下(扩展数据图1),
- 他们两个采样的BCR::ABL1阳性菌落(图2)与诊断时的BCR::ABL1阳性基因组相比,在CpG位点上C>T的比例较低(平均0.18)(图3f),
- 这表明尽管这些谱系携带着BCR::ABL1,但它们仍然以正常速率分裂。
- 我们确认这些菌落和诊断样本中的BCR::ABL1易位事件是相同的。
- 这一不寻常的观察提出了几种可能性:
- 一是TKI疗法导致仅快速分裂的BCR::ABL1突变细胞被优先清除,
- 二是TKI疗法减缓了某些BCR::ABL1阳性细胞的分裂速度,
- 三是BCR::ABL1的下游后果在这种患者的表观遗传上被沉默。
- 竞争性克隆造血或免疫调节也可能限制了剩余BCR::ABL1细胞的分裂速度。
Telomere lengths in CML
Para_01
- 端粒是位于染色体末端的重复DNA序列,随着细胞分裂而缩短,是细胞分裂历史的直接读出。
- 在健康的造血干细胞和祖细胞(HSPCs)中,端粒损耗已被证明每年约为30个碱基对,与这里观察到的野生型细胞相似(n = 469,每年-30.8个碱基对,CIboot -12.6至48.2个碱基对)。
- 模型截距提供了出生时估计的端粒长度,为6,180个碱基对(CIboot 5,216至7,086个碱基对),与之前的估算一致(9)。
- BCR::ABL1阳性的HSPCs(n = 365)尽管采样时年龄较轻,但端粒长度明显减少,额外损失了556.9个碱基对,且这一损耗独立于年龄(CI -86.0至993.2个碱基对,补充说明2和图3g)。
- 我们注意到PD51635 BCR::ABL1阴性的HSPCs中的端粒异常短小,这可能反映了野生型区室内克隆性造血的细胞分裂历史增加(图3h)。
Timing and rate of BCR::ABL1 expansion
Para_01
- 我们通过建模在克隆扩展开始前以恒定速率积累突变来计算BCR::ABL1克隆扩展的起始时间,之后突变的获取速率更高,如上所示。
- 共线性的时机是使用我们改进的适应于考虑在长‘树干’末端发生的转化事件的Markov链Monte Carlo方法rtreefit23推断出来的,该事件发生在克隆扩展之前(补充说明3)。
- 使用这些基于时间的树,我们接下来使用我们的phylofit7工具估计生长速率,该工具假设一个S形克隆生长曲线。
- 指数增长阶段的前期范围上限s增加,直到后验估计的s对进一步增加不敏感(扩展数据图5a)。
- 我们报告年化增长率S = es − 1,以百分比形式表示,并与瞬时每年增长率s一起报告,在一个小的时间段δt内,以年为单位测量,克隆大小的分数增加大约是sδt。
Para_02
- 在两名最年轻的患者(PD57334,22岁;PD56961,36岁)中,从克隆扩增的发作到临床诊断的时间仅为3.2年(置信区间CI 2.4–4.2年)和3.9年(置信区间CI 3–5.3年,图4a和补充表3和5)。
- 根据最近的肿瘤起源估算,增长速率非常高,分别为每年99,000,000%(置信区间CI 906,000–13,500,000,000%,s = 13.81,置信区间CI 9.11–18.72)和每年26,800%(置信区间CI 11,800–63,000%,s = 5.59,置信区间CI 4.78–6.44,图4b和补充表3和5)。
- 这相当于突变克隆群每18到45天翻一番。
- 三名中年患者(45-55岁,PD51632、PD51634、PD57335)也有近期和快速的BCR::ABL1克隆扩增至诊断,时间范围从4.4年(置信区间CI 3.4–5.8年)到6.3年(置信区间CI 5.0–8年),增长速率从每年73,500%(置信区间CI 24,000–240,000%,s = 6.60 置信区间CI 5.49–7.78,PD57335)到每年2,700%(置信区间CI 1,700–4,300%,s = 3.33 置信区间CI 2.89–3.79,PD51634)不等(图4a、b和补充表3和5)。
Fig. 4: Timing and fitness of BCR::ABL1-driven clonal expansion.
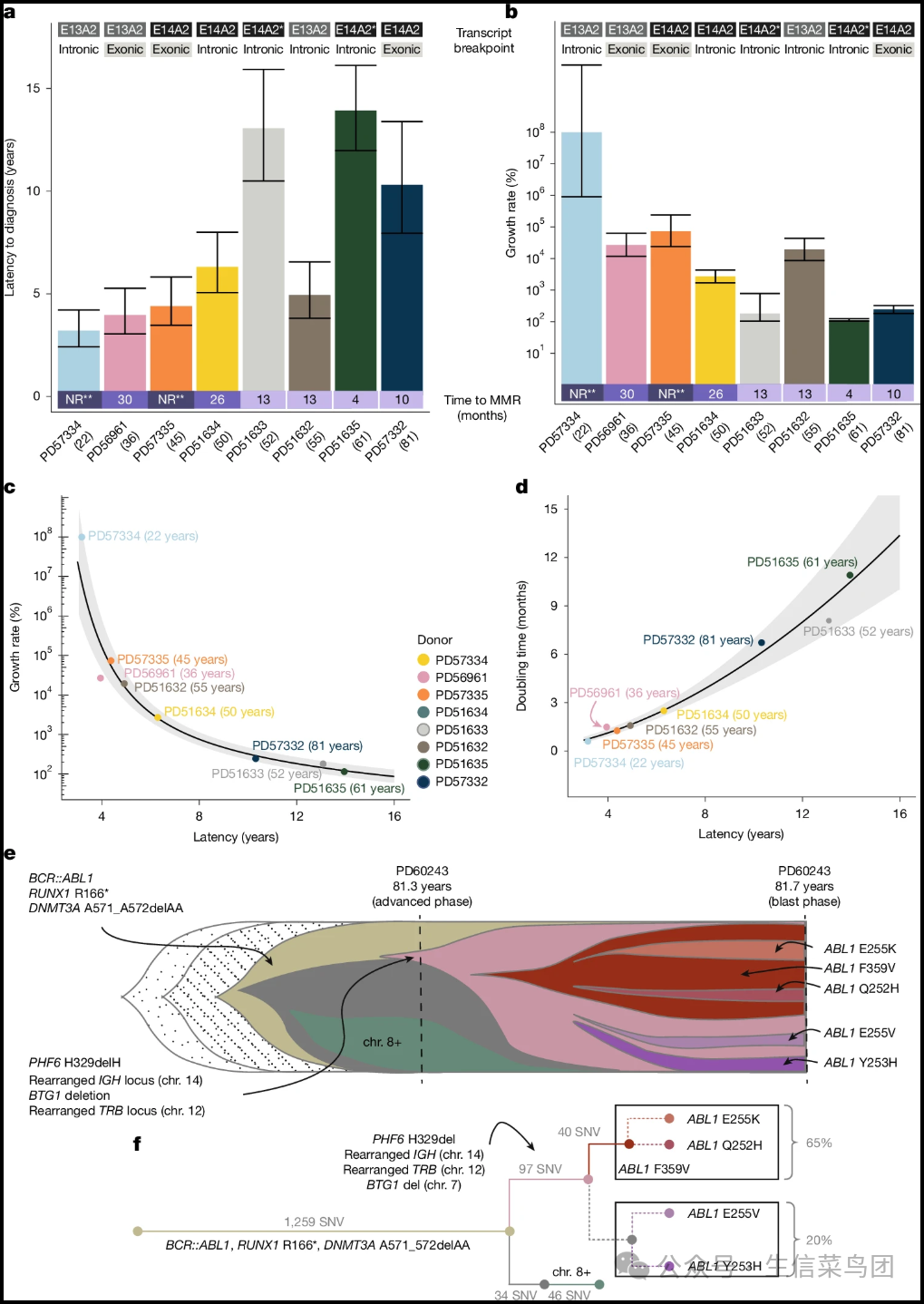
Fig__4__Timing_and_fitness_of_BCR__ABL1-_driven_clonal_expansion_.png
Fig__4__Timing_and_fitness_of_BCR__ABL1-_driven_clonal_expansion_.png
- 图片说明
◉ 条形图显示了基于rtreefit的潜伏期(以年为单位)(a)和基于phylofit的方法(方法)的年度化增长率(百分比)(b),患者按诊断时的年龄排序。◉ 条形图的高度和垂直线分别表示后验中位数和等尾95%可信区间。◉ 顶部的条带表示突变转录类型以及内含子与外显子非框内BCR和ABL1断点。◉ 星号*表示非互惠性BCR::ABL1易位伴有der(9)丢失。◉ 达到主要分子缓解(MMR)的时间(通过国际标准确定BCR::ABL1/ABL1比率小于0.1%)显示在下方。◉ NR表示未达到MMR。◉ **PD57334和PD57335的随访时间分别为23个月和21个月。◉ c,推断的BCR::ABL1获得(年)与每年BCR::ABL1阳性克隆的年度化增长率(%)之间的关系。◉ 点基于S和潜伏期的后验中位数估计,拟合线和95%置信区间(灰色带)来自线性模型log(log(1 + S)) ~ log(latency)。◉ d,估计的倍增时间(=1/log2(1+S))与从BCR::ABL1诱导的克隆扩增到诊断的时间('潜伏期')的关系。◉ 拟合线和95%置信区间(灰色带)来自c中使用的线性模型。◉ e,f,描述在PD60243中发现的晚期阶段(81.3年)和急变阶段(81.7年)的克隆广泛结构的鱼状图(e)和树状图(f)。◉ 通过基于读取的相位确认ABL1突变克隆存在于不同细胞中。◉ 树重建预测了中位数为2(范围1-3)的ABL1突变克隆嵌套在ABL1 p.F359V克隆内,一个例子如所示。◉ 鱼状图(e)y轴反映了每个测量时间点上推断的癌细胞比例,所示的克隆出现点仅用于说明。◉ 鱼状图(d)左侧的灰色部分(虚线和实线)代表具有单一(灰色虚线)和两个(灰色虚线)驱动突变的历史突变克隆:BCR::ABL1、RUNX1和DNMT3A;这些事件的顺序未知。◉ 树状图(f)展示了与e相同的树解决方案,并注释了相应亚克隆的常染色体SNV负担。◉ 虚线表示可能的分支结构。◉ 框用相应的克隆分数标记,对应于急变阶段样本PD60243d。◉ 来源数据
Para_03
- 相比之下,在三位年龄分别为52岁(PD51633)、61岁(PD51635)和81岁(PD57332)的患者中,我们观察到了稍长的克隆轨迹,分别为13.1年(置信区间10.5-16年)、14.0年(置信区间12.0-16.2年)和10.3年(置信区间7.9-13.4年)。
- 这些患者的克隆扩增速度处于中间水平,但仍然达到了每年114%-245%的增长(标准差为0.76-1.24,图4a、b所示)。值得注意的是,PD51635患者的生长速率起初似乎较慢(根据较长的共祖先区间判断),然后在一个亚克隆中变得更快(图2和补充表3、5所示)。
Para_04
- 鉴于如此快速的推断生长率,我们使用phylofit进行了基准测试,以评估是否可以准确推断出较大的生长率(补充说明3)。
- 使用cloneRate24('maxLikelihood'和'birthDeathMCMC')进行的正交生长率估计给出了相似的生长率估计(扩展数据图5b和补充表4),为CML中的非常高的估计生长率提供了额外的支持。
- 一个显著的例外是PD57335,在突变株系中后期合并对cloneRate估计有明显影响,但在使用phylofit时则没有(补充表4)。
- 正如之前报道的那样24,phylofit可信区间比cloneRate的更不保守。
Para_05
- 考虑到患者数量较少,似乎有一个合理的趋势,即发病年龄越小,与BCR::ABL1克隆扩增开始到诊断的时间较短以及增长更为迅猛相关(图4a、b),而年龄较大的个体则显示出相对较不迅猛的克隆扩增。
- PD51632是这些趋势的一个轻微异常值,因为在CML诊断前的8个月内曾接受过化疗治疗结直肠癌,这可能增强了对BCR::ABL1阳性HSPCs的选择。
Para_06
- 我们探讨了慢性髓性白血病(CML)生长速率差异的临床意义及临床反应。
- 队列中最快速生长的三种CML均未能在12个月内达到最佳分子缓解(根据国际标准定义为BCR-ABL1/ABL1比率小于1%),原因是TKI引起的细胞减少和BCR::ABL1水平降低缓慢(扩展数据图1)。
- 相比之下,在最年长患者中最慢生长的两种CML克隆却实现了快速的主要分子缓解。
- 这些观察结果提示,CML中的生长速率可能影响早期治疗反应。
Para_07
- 尽管在这小范围内观察到的临床表现模式各不相同,我们观察到 CML 的推断生长速率与临床表现时间之间有很高的相关性(图 4c、d)。
- 鉴于这两个参数是从树的不同特征中推断出来的,即克隆扩增开始时患者的年龄与克隆扩增内的合并模式(图 2 和方法部分),这种模式令人瞩目。
- 这一观察结果提供了证据,证明可以根据肿瘤生长速率预测临床表现,并表明在不可避免的临床表现之前,CML 白血病干细胞的人口规模有一个可以容忍的上限(补充说明 4)。
Disease progression in CML
Para_01
- 进展到慢性髓性白血病(CML)的爆发期与RUNX1、BCOR、TP53、ABL1、+8和等臂染色体17q(i(17q))中的额外突变有关。
- 配对的外显子组和目标基因测序在CML进展前后也显示突变负担增加。
- 我们通过外周血单核细胞或粒细胞DNA的全基因组测序,分析了四名年龄在38至81岁之间的晚期CML患者,这些患者的特征是循环中的原始细胞数量升高(10-19%)。
- 在所有病例中,我们观察到了BCR::ABL1克隆内的进一步驱动突变,例如RUNX1 p.Arg166、ASXL1 p.Tyr59fs、ASXL1 p.G646fs12和BCOR p.Leu532fs,以及拷贝数改变,如+1q、−16q、+8和i(17q)(补充表6)。
- 带有额外驱动因子的BCR::ABL1扩展,主导了CML克隆,几乎不留历史上的仅含有BCR::ABL1的克隆的证据。
- 这与本研究中的慢性期CML形成对比,在慢性期CML中,大多数情况下(八例中有六例),BCR::ABL1克隆内的额外驱动因子要么不存在,要么处于低水平。
- 在两个慢性期病例中,我们确实观察到了额外驱动突变的高克隆负荷:胎儿期NF1移码突变(PD51634)和RUNX1突变(PD57332)。
- PD51634随后发展为CML的爆发期,而PD57332在接受治疗后不久去世,疾病阶段尚不确定。
- 这些数据显示,晚期CML主要是由基因组驱动的,特征是获得额外驱动突变的BCR::ABL1克隆,这些突变提供了进一步的选择优势。
Para_02
- 一名患有晚期慢性髓性白血病(CML)(PD60243)的患者出现了急变。
- 这名81岁的患者在初次就诊时血液中异常细胞(11%的CD7+/CD13+/CD33+/CD117+/HLA-DR+细胞)数量增加,并且检测到RUNX1基因的p.Arg166*和DNMT3A基因的p.Ala571_Ala572del突变,此外还有BCR::ABL1融合基因(图4e,f)。
- 这个克隆体共享至少1,259个突变,与正常造血干细胞在这个年龄预期的突变数相差不大,表明该克隆体的最常见祖先(MRCA)是在相对较近的时间内形成的(可能是在人生的第七十年代)。
- 发现了两个亚克隆:(1)三体8号染色体(占BCR::ABL1/RUNX1/DNMT3A阳性细胞的56%)和(2)PHF6基因的框内p.H329delH突变(占BCR::ABL1/RUNX1/DNMT3A阳性细胞的14%)(图4e,f)。
- 在使用尼洛替尼治疗几个月后,患者的外周血中大约有20%的异常细胞,此时携带PHF6突变的亚克隆成为了主要的克隆。
- 该克隆显示出进一步的局部事件——BTG1基因缺失(12号染色体),TRB重排(7号染色体)和IgH重排(14号染色体)——这些都是典型的B细胞变化,符合淋巴样急变的特点。
- 这证实了淋巴样特征和分化是在通过RAG介导的基因组进化过程中,在获得BCR::ABL1之后出现的。
- 在这个高度突变的BCR::ABL1克隆体内,存在五种不同的ABL1突变亚克隆,其中ABL1.p.Phe359Val突变为主要突变(占细胞的65%)。
- 其他ABL1突变包括p.Q252H、p.Y253H、p.E255K和p.E255V。
- 我们的树状重建模型表明,1至3个携带ABL1突变的亚克隆可能在已经携带ABL1.p.Phe359Val突变的细胞中发生,这与在一个克隆体内可以共存多个ABL1突变的观点一致。
- 聚集在一起的ABL1突变亚克隆在急变期占细胞的70%-99%(图4e,f)。
- 亚克隆显示有数百个额外的全基因组突变,这与仅过去了一年零四个月的时间不符,表明早期的ABL1突变获得、快速的克隆生长和/或急变期的基因组不稳定性。
Discussion
Para_01
- 大多数癌症遵循多步骤的发展轨迹,随着克隆逐渐获得导致恶性肿瘤的遗传和表观遗传改变。这个过程通常始于几十年前,常常是30-50年,之后才会出现癌症症状。
- 相比之下,慢性髓性白血病(CML)是一种不同寻常的肿瘤。由单一获得的基因改变驱动的癌症很少见,并且我们没有发现除BCR::ABL1之外还需要其他共存的基因组改变来促进克隆扩展。
- ‘一次打击’癌症可以在儿科环境中观察到,例如婴儿期发生的MLL重排急性淋巴细胞白血病36和由单一基因融合驱动的儿童脑室内膜瘤37,38。
- 这些肿瘤可能发生在易感发育窗口期内。除了儿科环境外,由单一基因事件驱动的血液癌症,如由突变型JAK2驱动的骨髓增生性肿瘤,也表现出数十年缓慢的克隆生长7,9,32,39。
- 然而,成人CML的发展轨迹与此趋势相反,表现为爆炸性的快速肿瘤增长,肿瘤倍增时间仅为2-3周,临床疾病在3-4年后就会出现。
- 与之相符的是,我们的数据显示人群中不存在BCR::ABL1克隆性造血。
- 在BCR::ABL1驱动下随后的基因组演化促进了疾病的进展,这与之前的研究所述一致15,17,40,但也决定了在母细胞期的细胞身份。
Para_02
- 我们的估算表明BCR::ABL1克隆扩增到疾病表现的持续时间落在日本遭受原子弹轰炸幸存者的放射生物学研究中的癌症发病率范围内。
- 这表明电离辐射通过增加BCR::ABL1的启动来提高CML的发病率,并且随后的CML克隆轨迹忽略了BCR::ABL1初始获取的方式。
- 事实上,从BCR::ABL1获得到临床表现的如此短的时间只能通过极其快速的增长来解释。
- 我们对持续时间和生长速率的估算基于系统发育树的独立参数;具体来说,共享包含BCR::ABL1分支的长度以及克隆扩增期间共合区间模式。
- 我们使用正交数学建模验证了我们的估算,并观察到了通过加速突变获取和端粒损耗的独立证据,从而证实了快速生长。
- 因此,所呈现的数据提供了CML克隆动态的稳健推断。
Para_03
- 在我们的小型队列中,我们观察到生长速率的变化,其中三名年轻患者表现出爆炸性增长(每年超过10,000-1,000,000%),而两名年龄超过60岁的患者年增长率分别为114%和275%。
- 我们的队列规模太小,无法做出明确的相关性结论,但可以推测可能的原因。
- 年龄可能是通过细胞外因素影响的因素之一,例如,克隆性造血(PD51635)的竞争克隆可能会阻碍老年患者的BCR::ABL1克隆扩展。
- 细胞内在原因,如端粒缩短、与年龄相关的HSPC变化或异常的t(9;22)事件(例如,在PD51633和PD51635中观察到的ABR1::BCR丢失)也可能影响BCR::ABL1的效力。
- 生长速率的变化可能会影响疾病反应,因为五名生长速率最高的个体中有四名未能达到最佳治疗目标,这与慢性髓性白血病(CML)缓慢生长的情况相反。
- 长期以来,人们已经认识到BCR::ABL1水平在TKI治疗后下降的速度与长期缓解的可能性之间存在着复杂的联系(43,44);然而,导致BCR::ABL1动力学患者间差异的机制基础一直缺乏。
- 我们的数据显示,肿瘤生长速度可能是一个重要因素。
- 每五个患有CML的患者中就有一个在两种TKI疗法上失败(45),早期治疗失败与不良预后相关。
- 因此,更好地理解CML癌症轨迹的作用可以使治疗更早个性化,从而增强肿瘤缓解效果。
Methods
Patients and sample acquisition
患者和样本获取
Para_01
- 外周血和骨髓样本是从慢性期CML患者身上获取的。
- 我们选择了对一线TKI(PD51633、PD51632、PD51635和PD57332)有反应的患者,
- 对一线TKI没有反应的患者(PD57334、PD57335和PD57333),
- 以及那些反应较慢的患者(PD56961和PD51634)。
- 还包括了处于进展期CML的患者。
- 患者提供了书面知情同意书,同意将他们的样本用于研究。
- 选择患者是为了涵盖从诊断到不同治疗结果的各种年龄段。
- 该研究得到了NHS研究伦理委员会批准号05/MRE/44和18/EE/0199的覆盖。
In vitro expansion of single-cell-derived blood colonies
单细胞衍生血液集落的体外扩增
Para_01
- MNCs是从外周血或骨髓样本中使用Lymphoprep TM(Stem Cell Technologies)分离出来的。
- MNCs的单细胞悬液在半固体甲基纤维素基培养基MethoCultTM H4034 Optimum(Stem Cell Technologies)中培养了10-14天,如先前所述7。
- 单独挑选出的集落被溶解在45 μl的RLT缓冲液(Qiagen)中。
RT–PCR for BCR::ABL1 transcript
BCR::ABL1转录本的RT-PCR检测
Para_01
- 使用标准化的逆转录聚合酶链反应(RT–PCR)协议确定了患者PD56961、PD57332和PD57335的转录类型。
- 使用的引物是:BCR-e1-A:GACTGCAGCTCCAATGAGAAC(BCR外显子1),BCR-b1-A:GAAGTGTTTCAGAAGCTTCTCC(BCR外显子12-13)和ABL-a3-B:GTTTGGGCTTCACACCATTCC(ABL1外显子3)。
- 这产生了344 bp的PCR产物用于转录e13a2(6 bp(e12)+ 106 bp(e13)+ 175 bp(a2)+ 57 bp(a3)),以及419 bp的PCR产物用于转录e14a2(6 bp(e12)+ 106 bp(e13)+ 75 bp(e14)+ 175 bp(a2)+ 57 bp(a3))。
DNA library preparation, sequencing and read alignment
DNA文库制备、测序和读段比对
Para_01
- 10–20 μl 的裂解菌落悬液使用‘激光捕获显微切割活检’流程进行了 WGS 文库制备,该流程包含八个循环的 PCR。
- 这一流程能够从 150-200 个细胞的输入量中生成高复杂度的 WGS 文库。
- 那些生成的文库 DNA 含量超过 2 ng μl−1 的样本被用于使用 Illumina NovaSeq 6000 机器进行配对末端、150-bp 的 WGS 测序。
- 通过 BWA-MEM(Burrows-Wheeler 对齐器)算法将读取序列与人类参考基因组(GRCh38,NCBI)进行了比对。
Somatic mutation identification and filtering
体细胞突变识别和过滤
Para_01
- SNV 是通过将每个菌落与一个虚拟的不匹配样本(PD38Is_wgs)进行比较来识别的。
- CaVEMan 的运行参数设置为‘肿瘤中的正常污染’为零,并且肿瘤或正常拷贝数分别设置为五或二。
- 支持 SNV 的读取必须具有大于或等于 140 的 BWA-MEM 对齐评分中位数和软剪切碱基数中位数为 0。
- 为了适应这个定制的管道,应用了进一步过滤(https://github.com/MathijsSanders/SangerLCMFiltering)。
- 使用未匹配的正常样本意味着这个过程同时调用了体细胞和种系 SNV。
- 去除种系 SNV 和测序伪影需要进一步过滤。
- 正如在参考文献 7 中所述,我们使用跨菌落的汇总信息和匹配的种系 WGS 颊部样本的读取计数来确保那些可能存在于种系样本中的真正的体细胞变异也被识别出来。
- 这些变异可能是胚胎变异或者是由于肿瘤在正常组织中的污染造成的。
- 短插入和缺失(indels)是使用 cgpPindel 调用的,标准 WGS cgpPindel VCF 过滤器被应用,但 F018 Pindel 过滤器被禁用,因为它排除了深度小于十的位点。
- 通过与匹配的正常样本或同一患者的野生型菌落进行比较来识别拷贝数异常(CNA)。
- 然后对菌落的 SNV 和 indels 的并集进行了处理,并使用 VAFCorrect 在属于该个体的所有样本(包括菌落和颊部样本)中计算读取次数。
- 这使得在一个或多个菌落中检测到的突变可以在所有其他菌落中被识别,从而完全捕捉到来自单个患者的不同菌落之间的突变共享模式。
- 这生成了一个数据矩阵,其中包含支持每个突变的读取次数、该位点的测序深度以及该患者在每个菌落中的等位基因频率。
- Brass 和 GRIDSS 管道用于调用结构变异。
- cgpPindel 和 Brass 都使用 PDv38is_wgs 作为未匹配的正常样本。
Creating a genotype matrix
创建基因型矩阵
Para_01
- 每个样本中的每个位点的基因型为1(存在),0(不存在)或NA(未知)。
- 我们以深度敏感的方式推断了基因型。
- 如果位点是突变的,我们认为一个菌落在给定位点的观察到的突变读取计数MTR服从二项分布(n=深度,P=预期VAF);
- 如果位点是野生型,则认为MTR服从二项分布(n=深度,P=0.01)。
- 如果其中一个状态至少比另一个状态有20倍的可能性,则将基因型设置为两种可能状态中最有可能的那个。
- 否则,基因型被设置为缺失(NA)。
- 对于常染色体位点,预期VAF为0.5,但对于X染色体、Y染色体和CNA位点,它被设置为1/倍性。
- 对于杂合性丢失位点,如果原始基因型为0,则将其基因型覆盖并设置为缺失。
Phylogenetic tree topology
系统发育树拓扑结构
Para_01
- 我们使用最大简约法通过MPBoot52构建了系统发育树拓扑结构。
- 该方法旨在使达到分配给每个样本的所有突变所需的最少变化。
- MPBoot的输入是包含个体缺失值的二进制基因型矩阵。
- 这些数据被导出为多序列比对的Fasta文件,每列代表一个菌落,突变为A,野生型为T,缺失值为?。
- 使用的命令行是:mpboot -f-bb 1,000。
Donor with no wild-type colonies
没有野生型菌落的供体
Para_01
- 没有捕获到PD57332的野生型菌落。树的构建如上所述,仅突变分支存在。
- 推断了突变‘主干’分支的近似特征如下:(1)估计突变分支的持续时间为平均根尖SNV负荷除以队列平均BCR::ABL1 SNV突变率,(2)推断了主干的持续时间(突变分支样本高度时的年龄),(3)然后将分子时间内的主干长度定为(主干持续时间)×(队列野生型SNV突变率)+(出生时预期的突变负担)。
Driver annotation
驾驶员注释
Para_01
- 随着每个系统发育树分支被分配了SNVs和indels,有可能筛选所有分支是否存在潜在驱动突变。
- 为此目的,使用了一个先前7编制的基因列表(n = 35),这些基因常见于克隆性造血和骨髓增殖性疾病:ASXL1、BCOR、CALR、CBL、CSF3R、CUX1、DNMT3A、EZH2、GATA2、GNAS、GNB1、IDH1、IDH2、JAK2、KIT、KRAS、MLL3、MPL、NF1、NFE2、NRAS、PHF6、PPM1D、PTPN11、RB1、RUNX1、SETBP1、SF3B1、SRSF2、SH2B3、STAG2、TET2、TP53、U2AF1 和 ZRSR2,并加入了ABL1和BCR,总计37个基因。
- 带有已识别驱动突变的分支在系统发育树上用颜色突出显示。
- 根据Brass和GRIDSS结果在各个菌落中存在或不存在的情况,手动向分支添加了BCR::ABL1融合的注释。
Timing branches
定时分支
Para_01
- 鉴于体细胞突变随年龄呈线性积累,我们可以推断驱动突变在系统发育树中发生的时间点。
- 树顶部的分支包含在年轻时获得的突变,而较低位置的分支代表后来生活中发生的突变。
- 我们使用了我们之前开发的方法 rtreefit(https://github.com/nangalialab/rtreefit)来将分支长度用分子时间(即,突变数量)表示的树转换为分支长度用时间单位(年)表示的树。
- 简而言之,该方法联合拟合野生型和突变型的恒定突变率(即,每年累积的SNV数量),以及绝对时间分支长度,使用基于贝叶斯的个体树模型。
- 假设分配给分支的观察到的突变数服从泊松分布,平均值等于分支持续时间×敏感度×突变率,并且受根尖持续时间等于采样时年龄的约束。
- 此外,该方法通过假设胚胎发育期间突变率升高来考虑这一因素。
- 在运行 rtreefit 时,突变克隆群被定义为不包括主干,因此该方法假定BCR-ABL1是在主干末端获得的。
- 捐赠者PD57332没有野生型样本,因此添加了一个模拟野生型外群(分支长度等于群体野生型突变率的1000倍)。
- 这使得PD57332能够以与其他捐赠者类似的方式通过rtreefit进行处理。
- rtreefit算法运行了四个链,每个链20,000次迭代。
- 使用treemut以深度敏感的方式将突变分配给树,突变被硬分配到最高概率的分支(https://github.com/nangalialab/treemut)。
- 根据分支特异性SNV检测灵敏度调整分支长度,其中完全克隆SNV变异的检测灵敏度直接从每种菌落检测种系杂合SNV的灵敏度估计,并加上一个乘法校正以反映菌落的克隆性(VAF)。
- 在计算突变负荷和分支长度时,个体中存在的任何菌落中的CNAs在所有菌落中统一屏蔽,然后总体突变负荷按1−预期突变数在屏蔽区域的倒数进行缩放。
- 除SNV、插入缺失和BCR-ABL1融合外,菌落还显示了多种CNA事件。
- 这些事件被整理为存在于或不存在于每个菌落中的状态,从而得到类似于SNV和插入缺失获得的事件基因型向量。
- 一旦使用SNV基因型推断出树的拓扑结构,就识别出与事件基因型完全匹配的分支,并将事件分配给相应的分支。
Quality control of a phylogenetic tree topology
系统发育树拓扑的质量控制
Para_01
- 初始的质量评估步骤包括移除对CaVEMan体细胞突变检测敏感性低于60%的菌落。
- 也排除了可能被其他菌落细胞污染的菌落。
- 如果菌落不是单克隆的,则分配给其私有分支的SNV的平均VAF将低于50%。
- 为了确保样本是单克隆的,如果映射到其私有分支的SNV的VAF显著低于0.4,则排除该菌落。
- 如果分配给一个菌落的突变在非祖先分支中的VAF不为0,则也排除该菌落,因为这表明该菌落的细胞正在污染其他菌落。
Per sample BCR::ABL1 mutation status
每个样本的BCR::ABL1突变状态
Para_01
- 为了确定每个样本的BCR::ABL1突变状态,我们使用GRIDSS进行了‘联合’基因分型方法。
- 对于每位患者,Brass结构变异调用被审查,包括所有与BCR::ABL1融合事件相关的感兴趣事件,该事件包含一个核心‘目标’区域,包括ABL1和BCR的基因区域。
- 对于PD51635,我们加入了RBFOX3(染色体17),对于PD51633,我们纳入了der(9)上的大片段缺失。
- 对于每位患者,所有样本都在目标区域一起分析,以共同识别结构变异的证据。
- 每位患者的单个结果经过审查,并使用gGnome可视化(https://github.com/mskilab-org/gGnome),这允许重建复杂事件(如PD51635)患者的衍生染色体。
- 为了确定易位的后果,我们使用了GRASS(基因重排分析系统,https://github.com/cancerit/grass)。
- 在PD51635中,一个复杂的结构变异t(9;17)事件(倒位‘chr. 9 130776970GG]chr9:130786796]’和易位‘chr. 9 130777016T [chr17:79184121[ATT’)被简化为等效的单一易位事件(chr. 9: +:130777016,chr. 17: +:79184121,AT)进行注释(图1d和补充表2)。
Prediction of consequence of exonic BCR::ABL1 breakpoints
外显子BCR::ABL1断点的后果预测
Para_01
- 在三名患者(PD56961、PD57332和PD57335)中,我们在BCR外显子(分别是外显子14、15和15)中识别到了断裂点。
- 为了识别终止密码子,我们从断裂点开始读取框内序列,并根据转位事件中插入的碱基进行调整。
- 我们使用SpliceAI53预测了每个相应的融合序列的剪接概率。
- 使用GRCh38参考序列(BCR:22号染色体23170509-23328037,ABL1:9号染色体130825254-130897675)以及融合事件(BCR::ABL1)中的10kb侧翼序列和额外碱基重构了融合序列。
- 重构的融合序列和参考序列被用作‘自定义序列’脚本(https://github.com/Illumina/SpliceAI)的输入。
- SpliceAI产生的‘原始’剪接受体和供体概率被转换为bedGraph格式并在IGV54上进行审查。
Mutational signature analysis
突变特征分析
Para_01
- 新生突变特征提取使用HDP(https://github.com/nicolaroberts/hdp)进行,其中分配到各个分支的突变被视作样本。
- 少于50个突变的分支被分为每供体两个一组;那些在分子时间100个突变前结束的短分支被归入‘生命早期’组,其余短分支则归入‘生命晚期’组。
- 这些组也被视作样本。
- 随后,HDP提取在四个链上运行,参数为:burnin=10,000,n=500,spacing=250。
- SBSblood特征55被下载并与泛癌全基因组分析特征合并(https://cog.sanger.ac.uk/cosmic-signatures-production/documents/COSMIC_v3.3.1_SBS_GRCh38.txt)。
- 新生提取鉴定出三个特征SBSblood、SBS1和SBS18,它们与其相应的已发表的泛癌全基因组分析和/或SBSblood特征的余弦相似度分别为0.927、0.942和0.884。
- 我们针对上述已发表的SBSblood、SBS1和SBS18版本对每个供体分支分组及单个分支进行了重新拟合。
- 这种特征分配是通过sigfit::fit_to_signature使用默认的'multinomial'模型对每个此类供体类别或分支进行的。
- 然后按以下步骤进行每个分支的特征分配:(1) 分配每个突变的特征成员概率,然后(2) 将所有分配给该分支的所有SNV的特征成员概率相加,以获得分支级别的特征分配比例。
- 每个突变的特征成员概率使用以下方法计算:
Para_02
- 先验概率 P0(Sig) 由属于该突变所属类别的指定签名 Sig 的平均 Sigfit 归属概率给出。
- 通过使用 'metafor' R 包中的 rma 函数,对分支类别中的突变特征和 CpG 位置的 C>T 表示进行了队列水平分析。
Growth rate estimation
增长率估算
Para_01
- BCR-ABL1 克隆的生长速率使用之前描述的 phylofit 方法进行估算7,9。
- 简而言之,phylofit 是一种贝叶斯方法,通过直接拟合由给定种群规模轨迹下的共祖先时间联合概率密度得出的三参数逻辑增长曲线轨迹来估计生长速率。
- 这种方法估计的三个参数是饱和种群规模 N、指数增长阶段的增长率 s 和曲线的中点 t(m)。
- 鉴于慢性髓性白血病患者表现出较高的 BCR::ABL1 比例,我们将中点先验的上限设定为诊断时的年龄,下限设定为突变克隆的最近共同祖先的年龄。
- 我们对增长率 s 以及饱和种群规模的对数尺度采用了均匀先验。
- 现在,年化增长率由 S = exp(s) − 1 给出,对于 s、中点 t(m) 和种群规模 N 的均匀先验分别是:
Para_02
- 选择 t(m) 的上限是基于这样的观察:慢性髓性白血病患者在诊断时通常表现出较高的 BCR-ABL1 负担。
- phylofit 的输入数据是从 rtreefit 获得的时间树,如上所述。
- 请注意,输入树的分支长度被选择为后验分支长度的平均值。
- 这些树被限制在最早的采样点。
- 这些时间点都在诊断时或诊断之后。
- 使用最近发表的替代方法 cloneRate24 进行了估计比较的检查,详情见补充说明 3。
Telomere analysis
端粒分析
Para_01
- Telomerecat (v.4.0.2, https://github.com/cancerit/telomerecat) 被用来估计平均端粒长度(以碱基对为单位),使用了 (-t 75) 来减轻NovaSeq测序技术带来的影响(补充说明详见补充说明2)。
Mixed models
混合模型
Para_01
- 用于SNV负荷和端粒分析的线性混合模型通过R包lme4实现,以估计年龄和突变状态的影响。
- 年龄(age_at_sample_exact)被定义为取样时从出生到完成年数的计数,样本突变状态(BCR_ABL1)被定义为BCR::ABL1阳性(Mt)或阴性(Wt)。
- 对于每个响应变量(y),我们首先测试突变状态的显著性(模型0对比模型1),然后进一步的项被加入以查看这是否改善了与基础模型(模型1)相比的模型。
Para_02
- 模型使用了 lme4 包的默认参数进行拟合。如果模型没有收敛,使用 lme4::allFit() 函数重新拟合模型到所有可用的优化器(由 lme4、optimx 和 dfoptim R 包提供),从非奇异且收敛的重新拟合中选择具有最高负对数似然的最佳优化器。
- 只有非奇异且收敛的模型被考虑用于使用贝叶斯信息准则(BIC)进行的模型选择。
- 对于最终选定的模型,使用 lmeresampler R 包中的 bootstrap() 函数生成的前 1,000 个收敛且非奇异的参数自助重抽样模型中的 confint(type='perc') 函数计算每个固定效应的 95% 置信区间(百分位自助区间),以确保可重复性,使用种子值 1234。
SNV burden models
SNV负荷模型
Para_01
- 我们首先移除了所有来自PD57332的样本(n = 35),因为我们只能培养出BCR::ABL1菌落,因此我们预计对SNV突变负荷(nsub_adj)的估计会存在偏差;这使得剩下的样本数为799个,涉及八名患者。
- 最终使用的模型如下面所示,并且发现它具有最低的BIC值(8,645.52),详细信息见补充说明1。
Telomere length models
端粒长度模型
Para_01
- 为了应对使用 Telomerecat 和 NovaSeq 测序数据进行端粒估计时报告的问题9,我们在野生型样本(n = 469)中探讨了文库制备(library.cluster)和测序运行(run_id.uniq)中的任何批次效应对平均端粒长度(length)的影响。
- 将测序运行作为额外的随机效应(1| run_id.uniq)加入模型后,得到了具有最低 BIC 值(7,483.33)的模型。
- 这一项被加入了一系列基于完整数据集(n = 834)测试的模型中,最终模型具有最低的 BIC 值(13,265.15),具体细节见补充说明2。
Bulk DNA library preparation, sequencing and read alignment
大规模DNA文库制备、测序和读段比对
Para_01
- 从外周血提取的DNA进行了全基因组测序文库制备,并使用Illumina NovaSeq 6000机器进行了配对末端测序,读长为150 bp。
- 使用BWA-MEM算法将读段比对到人类参考基因组(GRCh38,NCBI)上。
Bulk unmatched somatic mutation identification and filtering
批量识别和过滤大规模非匹配体细胞突变
Para_01
- SNVs是通过将每个bulk样本与一个虚拟的不匹配样本(PD38Is_wgs)进行比较来识别的。
- CaVEMan运行时将‘肿瘤中的正常污染’参数设置为零,并且将肿瘤和正常拷贝数分别设置为五和二。
- 为了提高敏感性,只在正常样本面板中被标记为‘VUM’的SNVs被恢复。
- 所有SNVs必须具有少于一半的支持读段剪切(CLPM=0),并且中位BWA-MEM对齐得分大于或等于140(ASMD≥140)。
- 短插入和删除(indels)是使用cgpPindel48调用的,并应用了标准WGS cgpPindel VCF过滤器,但排除了F010 Pindel过滤器,因为它排除了在正常样本面板中看到的变异。
- 驱动候选变异限制在上述描述的37个基因内。
- 为了过滤种系变异,我们保留了gnomAD v.3.1.2(参考文献56)的最大人群等位基因频率小于0.01的SNVs和indels(使用echtvar v.0.2.0注释,参考文献57)。
- 合并了SNVs和indels,并使用VAFCorrect在所有属于该个体的样本中计算了读段。
- 通过IGV54审查测序读取深度,以识别臂级拷贝数事件。
- GRIDSS v.2.13.1用于结构变异调用,并估计BCR::ABL1的VAF。
Bulk phylogeny reconstruction of PD60243
PD60243的大规模系统发育重建
Data availability
Para_01
- 测序文件可通过欧洲基因组档案(EGA,数据集EGAD00001015353)获取,这符合韦尔科姆桑格研究所的数据共享政策,并且所有体细胞突变.vcf文件已上传至Mendeley(https://doi.org/10.17632/yg29vx2f35.1)。
- 通过研究者工作台,全世界的研究人员可以使用个体层面的数据进行研究,该工作台是基于云的计算平台(https://www.researchallofus.org/register/)。
- 汇总层面的数据可以通过由研究项目提供的数据浏览器向公众开放(https://databrowser.researchallofus.org/)。
- 本论文提供了源数据。
Code availability
Para_01
- 为分析创建的代码和软件可在GitHub (https://github.com/nangalialab/CML) 获取。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-05-08,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号