三代测序人物系列 | Yi Xing (邢毅)
原创

三代全长转录组可变剪切分析相关软件:rMATs-long, l2rmats和 espresso。二代RNA-seq测序经典可变剪切事件分析软件 rMATs 和 rmats-turbo 都出自邢毅(Yi Xing)教授的团队。
邢毅(Yi Xing)教授

邢毅(Yi Xing)教授
美国费城儿童医院(Children’s Hospital of Philadelphia ,CHOP)计算与基因组医学中心(Center for Computational and Genomic Medicine)的创始主任,以及生物医学与健康信息学系的执行主任,同时还担任宾夕法尼亚大学(University of Pennsylvania)病理学与实验医学系(Department of Pathology and Laboratory Medicine, Perelman School of Medicine,)的教授。在加入CHOP和宾大之前,邢博士曾是加州大学洛杉矶分校(UCLA)微生物学、免疫学和分子遗传学系的教授。
邢毅(Yi Xing)教授团队在生物信息学、基因组学和RNA生物学领域发表了大量论文,他的研究为哺乳动物转录后RNA加工的功能、调控和进化提供了基础性见解。他目前的研究结合了计算生物学、生物医学数据科学、RNA基因组学、人类遗传学、精准医学和免疫肿瘤学等领域的知识。
实验室主页: https://xinglab.org/

邢毅教授实验室成员
一、实验室研究方向
邢毅(Yi Xing)教授团队研究的长期目标是阐明哺乳动物转录组和蛋白质组中的可变异构体(alternative isoform)复杂性。广泛关注RNA加工和调控的计算生物学和基因组学,以及它们在人类遗传学、精准医学和癌症免疫治疗中的应用。
哺乳动物细胞从数量较小的基因集合中产生了令人惊叹的调控多样性和复杂的表型。我们现在知道,这些的多样性是通过RNA的可变剪切(alternative splicing)和修饰(modifications )实现的。我们研究的长期目标是阐明哺乳动物转录组和蛋白质组中的可变异构体(alternative isoform)复杂性,理解它们是如何产生的,以及它们在复杂基因组调控和功能中的作用。邢毅(Yi Xing)教授团队开发了用于研究组织(bulk)/单细胞(single-cell)样本转录组和蛋白质组复杂性的计算方法和基因组技术(Nature Methods, 2019; American Journal of Human Genetics, 2019; Genome Biology, 2017; Nature Methods, 2016; PNAS, 2014; Genome Biology, 2013)。还整合计算和基因组工具,以阐明健康和疾病中的RNA调控网络(American Journal of Human Genetics, 2018; Genome Biology, 2016; Cell Reports, 2016; Elife, 2015; Cell Stem Cell, 2014; Molecular Cell, 2014; Neuron, 2014)。
目前活跃的研究方向和课题包括但不限于:
- 转录组分析相关算法的开发(第二代和第三代测序数据)。
- 关于分析RNA加工和修饰技术和算法的开发 (针对痕量RNA和单细胞样本)。
- 利用大规模RNA-seq数据和蛋白质-RNA互作图谱研究健康和疾病中的RNA调控网络。
- 转录组调控和RNA加工的遗传变异与进化。
- 用于疾病诊断或早期检测的临床RNA-seq技术。
- 精准肿瘤学和癌症免疫治疗 (利用多组学和临床数据整合)。
二、开发软件
1. rMATS-long
rMATS-long 是一个用于长读长RNA测序数据的计算工作流程。基于作者团队开发的另一款用于长读长RNA测序数据转录本发现和定量的工具Espresso,rMATS-long能够实现样本组之间的差异异构体( isoform)分析,以及异构体( isoform)结构和丰度的分类与可视化。
软件安装
可以使用mamba或者conda安装
$ conda install -c conda-forge -c bioconda rmats-long
# 或
$ mamba install -c conda-forge -c bioconda rmats-long分析步骤
- Detect differential isoform usage: scripts/detect_differential_isoforms.py
- Visualize isoforms and abundance: scripts/visualize_isoforms.py
- Classify isoform differences: scripts/classify_isoform_differences.py
2. ESPRESSO
Espresso(Error Statistics PRomoted Evaluator of Splice Site Options)是一种新的,用于处理比对后的长读长RNA测序数据的方法,能够有效提高识别剪接区域的准确性以及异构体(isoform)定量的精度。
软件安装
可以使用mamba或者conda安装
$ conda install -c bioconda espresso
# 或
$ mamba install -c bioconda espresso分析步骤
# 示例
$ perl ESPRESSO_S.pl -L samples.tsv -F ref.fasta -A anno.gtf -O work_dir
$ perl ESPRESSO_C.pl -I work_dir -F ref.fasta -X 0
$ perl ESPRESSO_Q.pl -L work_dir/samples.tsv.updated -A anno.gtf3. rMATS
rMATS是一款对RNA-Seq数据进行差异可变剪切分析的软件。其通过rMATS统计模型对不同样本(有生物学重复的)进行可变剪切事件的表达定量,然后以likelihood-ratio test计算P value来表示两组样品在IncLevel(Inclusion Level)水平上的差异。
MATS可识别的可变剪切事件有5种:
- skipped exon (SE) 外显子跳跃
- alternative 5' splice site (A5SS) 第一个外显子可变剪切
- alternative 3' splice site (A3SS) 最后一个外显子可变剪切
- mutually exclusive exons (MXE) 外显子选择性跳跃
- retained intron (RI) 内含子滞留
4. rmats2sashimiplot
rmats2sashimiplot 是一个用于可视化 rMATS 输出结果的工具。它还可以通过注释文件和基因组坐标生成绘图。该工具的绘图后端是 MISO(Mixture of Isoforms)。
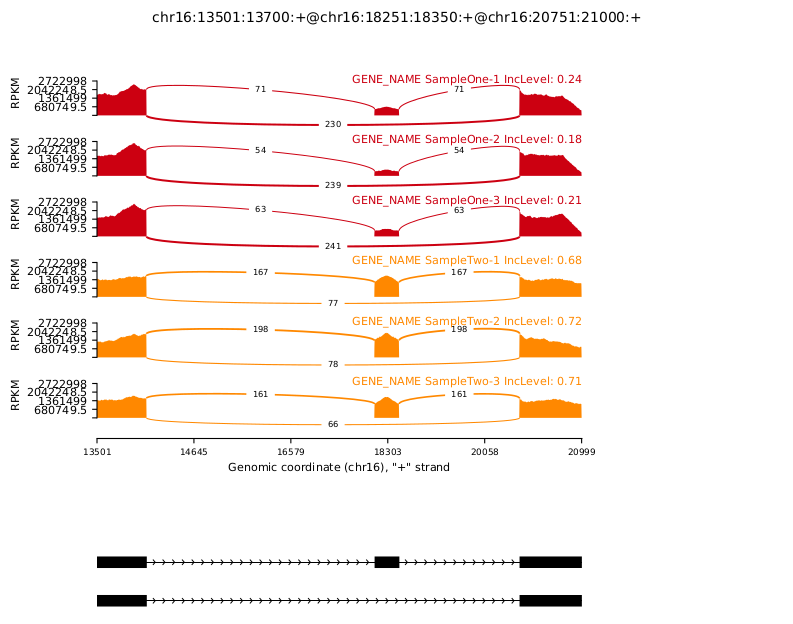
sashimiplot示例图
参考文献
- Xinglab:https://xinglab.org
- Github: https://github.com/Xinglab
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号