Vitessce: 多模态和空间分辨单细胞数据的综合可视化



Basic Information
- 英文标题: Vitessce: integrative visualization of multimodal and spatially resolved single-cell data
- 中文标题:Vitessce: 多模态和空间分辨单细胞数据的综合可视化
- 发表日期:27 September 2024
- 文章类型:Brief Communication
- 所属期刊:Nature Methods
- 文章作者:Mark S. Keller | Nils Gehlenborg
- 文章链接:https://www.nature.com/articles/s41592-024-02436-x
Abstract
Para_01
- 多组学技术与单细胞和空间分辨率相结合,使得测量数百万个细胞中的数千种特征成为可能。
- 然而,同时对高维转录组学、蛋白质组学、基因组映射和成像数据类型进行视觉分析仍然是一项挑战。
- 在这里,我们描述了Vitessce,一个用于探索多模态和空间分辨单细胞数据的交互式基于网络的可视化框架。
- 我们展示了在多个协调视图中整合可视化数百万个数据点的能力,包括细胞类型注释、基因表达量、空间分辨转录物和细胞分割。
- 该开源软件可在http://vitessce.io获取。
Main
Para_01
- 技术手段已经克服了组织和器官在单细胞分辨率下空间定位和多模态测量的挑战,提高了我们区分细胞类型、细胞状态和细胞邻域的能力。
- 单细胞组学和成像数据的高维度促进了计算模型的发展,这些模型旨在通过分割、聚类成员身份和预测的细胞类型来注释细胞观察。
- 对整合了基因组学、转录组学和成像模态的多模态和空间分辨的单细胞数据集进行视觉分析可以促进质量控制、结果交流、生物标志物识别以及假设的生成。
Para_02
- 尽管该领域在实验和计算方面取得了进展,但仍缺乏可扩展的工具来交互式地可视化多模态和空间单细胞数据集。
- 可视化研究社区已经指出了对异构生物数据集进行视觉分析的工具不足问题,这一问题被称为‘超大规模综合可视组学的挑战’2,3。
- 目前,多模态单细胞数据集的可视化需要多个不相关的工具,如图像查看器和基因组浏览器,它们支持单一的数据模态。
- 有助于跨模态发现连接的工具将使多模态实验数据能够充分发挥其潜力,以提高对健康、疾病和调控机制的理解4。
Para_03
- 最近用于单细胞数据可视化的工具侧重于交互性和可扩展性。
- Cellxgene5、Cirrocumulus6 和 Pagoda27 等工具支持使用散点图和热图对包含数百万个细胞和数千个基因的数据集进行交互式探索。
- 然而,这些工具是为转录组学数据的视觉分析而设计的。
- 因此,它们缺乏对空间数据类型(如细胞分割)或基因组映射模态(如染色质可及性谱)的支持。
- 另一方面,TissUUmaps8 和 Napari-SpatialData9 等针对空间数据的工具不支持非空间或基因组映射数据可视化。
- 迄今为止,单模态数据的重点导致了无法支持跨模态比较和关系以及空间上下文的可视化任务的工具。
Para_04
- 我们开发了 Vitessce 以克服这些限制,并实现多模态和空间单细胞数据的综合可视化。
- Vitessce 是一个可扩展的基于网络的数据可视化框架,支持同时对转录组学、蛋白质组学、基因组图谱和成像数据进行可视化探索(图 1)。
- 例如,该软件提供了基于 Viv11 和 HiGlass12 工具包的模块,分别用于多尺度多重成像数据和基因组图谱数据的可视化(图 2)。
- Vitessce 的一个优势是,它能够在各种情境和计算基础设施中部署空间和单细胞数据的可视化,包括静态网站、Web 应用程序、数据门户、Jupyter Notebook、RStudio 和 R Shiny 应用程序。
Fig. 1: Vitessce can be used in multiple settings and can be configured to visualize raw and derived measurements from multimodal and spatial single-cell experiments.
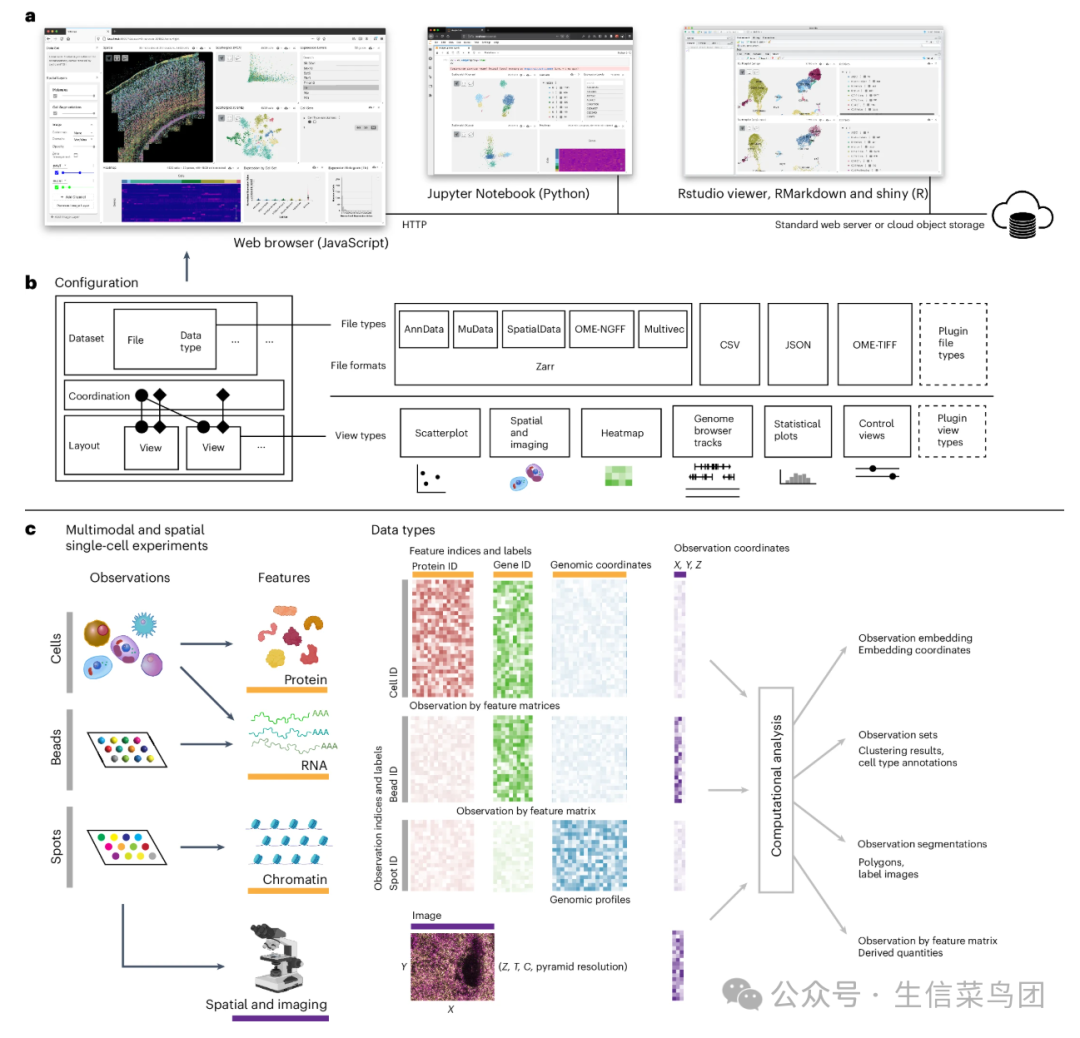
- Vitessce 可以作为网页浏览器中的一个 JavaScript 组件或 Python 和 R 分析环境中的一个小部件。
- 可以将存储在多种格式中的单细胞、单分子和显微镜数据可视化为多种类型的视图(即,交互式可视化)。
- Vitessce 的模块化设计使得能够整合地可视化多模态和空间单细胞实验与计算分析结果。
- 观察和特征之间的箭头表示能够可视化来自异构实验的数据,这些实验测量了部分共享于部分观察中的特征子集。
- OME(开放显微镜环境);NGFF(下一代文件格式);CSV(逗号分隔值);JSON(JavaScript 对象表示法);TIFF(标签图像文件格式);ID(标识符)。
- 图标来源说明:‘Observations’下的细胞图像来自 Reactome.org 并采用 CC BY 4.0 许可;‘Features’下的图像来自 https://www.biorender.com/ 。
Fig. 2: Sample use cases for Vitessce.
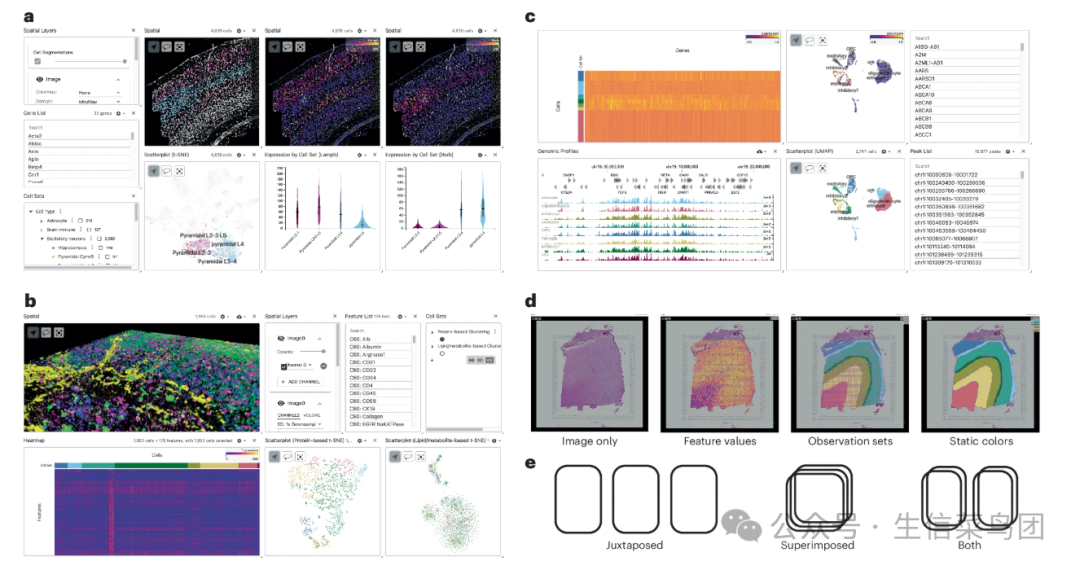
- 可视化包含多重图像、空间分辨RNA分子和细胞分割的单分子FISH实验。
- 三维多模态成像质谱(IMS)数据集的体积渲染,旁边附有热图以及基于蛋白质和基于脂质/代谢物的降维散点图的并排展示。
- 来自10x Genomics Multiome数据集的基因表达和染色质可及性的同时可视化。
- Vitessce空间视图中支持的可视化层示意图,即点、斑点、分割和图像。
- 斑点层和分割层可以根据特征值、集合成员或静态颜色进行着色。
- Vitessce支持多个空间分辨率可视化并置和叠加的排列方式。
- Chr代表染色体;t-SNE代表t分布随机邻居嵌入;UMAP代表一致流形逼近与投影。
Para_05
- 来自单细胞RNA测序(scRNA-seq)13和通过转座酶可及染色质测序(scATAC-seq)14的数据,以及多模态和空间技术如转录组和表位测序索引(CITE-seq)15、索引共检测(CODEX)16、Slide-seq17、10x Genomics Visium和Multiome可以使用Vitessce进行可视化。
- 必须在Vitessce之外或通过插件执行数据(预)处理(例如,降维和细胞分割)18,将可视化与生物信息学算法分离。
- Vitessce可以访问存储在静态web服务器和云对象存储中的文件,支持AnnData19,20、MuData21、SpatialData9、逗号分隔值(CSV)、开放式显微镜环境标记图像文件格式(OME-TIFF)22,23和OME-Zarr24,25格式。
Para_06
背景: 随着单细胞和空间分析技术的快速发展,Vitessce 框架旨在为大量 可视化生产者和使用者 提供服务。
Vitessce 主要面向:
- 需要与合作者交流研究结果的 单细胞数据分析人员。
- 希望探索静态图表之外数据的 科学出版物读者。
- 在后续研究前检查数据质量的 单细胞数据库用户。
Vitessce 解决了以下 五个关键挑战,使研究人员能够 构建、部署和访问 多模式与空间解析的单细胞数据可视化。
1. 根据数据特点定制可视化 挑战: 单细胞数据集可能包含多个模式(如基因表达、染色质可及性等),并可能带有空间坐标或图像。不同实验方法会生成多种数据类型,如降维结果、聚类、细胞类型注释和细胞分割等。
- Vitessce 支持 独立可视化不同数据模式,或将多模式数据投影到共享低维空间,以增强整合分析。
- 提供 模块化交互式视图,可用于高通量成像、细胞表达矩阵、细胞分割等多种数据类型。
- 采用 WebGL 和 deck.gl 处理大规模数据,如百万级细胞散点图、数万个特征的热图。
- 结合 Viv、HiGlass、Vega-Lite,支持 大规模图像、基因组测量及统计分析。
2. 通过多视图整合和探索数据 挑战: 现有的基因组浏览器、散点图工具、图像浏览器等相互独立,难以统一分析多模式数据。
- Vitessce 允许 协调多个视图,使得基因和细胞类型选择可在多个可视化视图中同步更新。
- 提供 50+ 配置参数,用户可自定义视图间的链接方式,实现高自由度的数据探索。
- 交互状态可保存和共享,例如:
- 通过 URL 共享可视化快照,确保结果可复现。
- 通过 配置文件 记录用户调整的参数,便于追踪分析过程。
3. 在不同计算环境中使用 Vitessce 挑战: 数据分析不仅限于 Web 浏览器,还涉及 Jupyter Notebook、R 语言等环境。
- Vitessce 可直接嵌入 Jupyter Notebook 和 R,作为交互式小部件,与代码共同展示分析结果。
- 提供 Python / R API,兼容 Scanpy、SnapATAC、Seurat 等流行单细胞数据分析工具。
- 支持 远程数据访问,可直接从云端存储或计算集群中加载数据。
4. 部署和共享交互式可视化 挑战: 传统交互式可视化工具往往依赖后端服务器,导致部署和共享较为复杂。
- Vitessce 采用 纯客户端架构,无需专门服务器,即可在浏览器端运行。
- 支持 Google Cloud Storage、AWS S3 等云存储的数据加载,无需额外后端服务。
- 提供 插件视图,支持与服务器端通信,实现更复杂的数据计算。
- 例如,Cheng 等人 研究中的插件视图,可与服务器交互,实现人机协作的数据整合。
5. 支持多种文件格式 挑战: 单细胞测量和显微镜数据缺乏统一的文件格式,不同实验和编程语言使用的格式各不相同。
- Vitessce 通过数据抽象层支持多种文件格式:
- 常见格式包括 CSV、AnnData、MuData、SpatialData、Multivec、JSON、OME-TIFF、OME-Zarr 等。
- 允许 插件数据加载模块 扩展新文件格式的支持,以满足不同实验需求。
结论: Vitessce 框架为 单细胞和空间数据可视化 提供了 高扩展性和易用性,使得数据分析人员、出版物读者、数据库用户都能 轻松探索、整合和共享数据。
未来展望: 随着单细胞技术的不断发展,Vitessce 可能会进一步扩展,支持更多计算环境、更大规模的数据,以及更丰富的交互模式。
Para_07
- Vitessce 能够实现对单细胞实验的可视化探索,这些实验涵盖了转录组学、表观基因组学、蛋白质组学和成像技术等多种模态,并在一个单一的集成工具中进行。
- 我们通过图2以及扩展数据图1-7中的用例展示了可视化构建、部署和访问的过程。
Para_08
- 在使用 CITE 测序的情况下,Vitessce 可用于验证由 RNA 和蛋白质模式中的标记物表征的细胞类型的存在。
- Stoeckius 等人15测量了人类脐带血单核细胞样本中的基因表达和表面蛋白丰度,研究了这些模式中的已知免疫细胞类型标记。
- 通过使用 Vitessce 可视化该数据集,可以重现 Stoeckius 等人的发现,使用链接散点图和热图同时探索蛋白质丰度和基因表达水平(扩展数据图5)。
- 例如,正如作者所报道的那样15,我们观察到可以通过基于 CD56 蛋白水平以及 GZMB、GZMK 和 PRF1 基因的表达来识别自然杀伤细胞。
Para_09
- 第二个使用案例展示了从单分子荧光原位杂交实验中可视化空间解析的基因表达数据。
- 在小鼠脑样本中,Codeluppi 等人发现了一种位于锥体 L2/3 和 L4 神经元细胞类型之间的过渡区域,该区域包含锥体 L3/4 细胞。
- 这一发现可以在 Vitessce 中通过空间图和热图视图重现(图 2a)。
- 在 Vitessce 中,细胞分割和散点图点的颜色可以映射到基因表达值。
- 通过交互选择基因 Lamp5 和 Rorb,我们可以观察到特定于锥体 L3/4 细胞的报道共表达模式。
- Vitessce的发展得到了细胞图谱联盟的合作推动。
- 该框架在人类生物分子图谱计划(HuBMAP)和肾脏精准医学项目(KPMP)的数据门户中的使用,以及其在GitHub上的开源开发,已经带来了宝贵的反馈,这些反馈指导了需求和未来的研究方向。
- Vitessce未来的发展方向包括支持比较分析、提高可扩展性以及为三维组织地图和基因组映射数据增加更多的可视化类型。
- Vitessce 脱离了任何特定的数据库,可以作为可视化前端集成到具有不同后端数据组织策略的项目中。
- 该框架的模块化和可扩展性允许支持随着发展而出现的其他可视化类型、文件格式和单细胞模式。
- 我们预计社区将通过贡献插件模块并将其纳入提供针对特定受众、任务和技术的便利的应用程序中来接受这些特性。
Methods
Using Vitessce
使用 Vitessce
Para_01
- Vitessce 以不同的形式存在,以便为多个受众群体提供便利。
- 研究人员可以在数据分析过程中使用 R 和 Python 包来探索本地或远程数据集。
- Vitessce 网站和在线配置编辑器旨在与合作伙伴共享可视化内容以及调试。
- 软件开发人员可以通过将其作为 JavaScript 包使用,将 Vitessce 集成到其他工具中并编写插件。
Vitessce in Python environments and Jupyter Notebooks
Vitessce 在 Python 环境和 Jupyter 笔记本中
Para_01
- Vitessce 可以作为 Python 包在脚本和 Jupyter 笔记本中使用。
- 可视化可以通过一个与多种笔记本环境兼容的交互式小部件直接配置和渲染到笔记本中,包括 JupyterLab、Jupyter 笔记本(经典版)和 Google Colab。
- 小部件的实现基于 AnyWidget(https://anywidget.dev)。
- 安装说明和 API 文档可以在 https://python-docs.vitessce.io 查找。
- 一套教程笔记本可以在 https://github.com/vitessce/vitessce-python-tutorial 获取。
Vitessce in R environments, R, RStudio and Shiny apps
在 R 环境、R、RStudio 和 Shiny 应用中使用 Vitessce
Para_01
- Vitessce 可以作为 R 语言包在脚本、R 文档和 Shiny 应用中使用。
- 在 R 中配置的可视化可以通过交互式小部件在 RStudio 查看器窗格、R 文档、pkgdown 网站和 Shiny 应用中呈现。
- 该小部件是作为 R htmlwidget 实现的。
- 安装说明和 API 文档可以在 https://r-docs.vitessce.io 查找。
Vitessce in JavaScript and web environments
Vitessce 在JavaScript和web环境中
Para_01
- Vitessce 是用 JavaScript 实现的,作为一个 React 组件,并且有相应的 API 用于配置和注册插件(如下所述)。
- JavaScript 包可以在网站、其他 React 组件或其他 JavaScript 包中使用。
- 安装说明和 API 文档可以在 http://vitessce.io 查找。
Vitessce as a website and online configuration editor
Vitessce作为一个网站和在线配置编辑器
Para_01
- 为了从网络浏览器快速配置 Vitessce 可视化,我们提供了一个网络应用程序,用于使用 JavaScript 对象表示法(JSON)和 JavaScript 语法编写和编辑 Vitessce 配置。
- 这种方法配置 Vitessce 实例不需要安装 Python、R 或 JavaScript 包。
- 此资源可以在 http://vitessce.io 找到。
Python, R and JavaScript configuration APIs
Python、R 和 JavaScript 的配置 API
Para_01
- Vitessce 配置是使用一种 JSON 表示法定义的,该表示法指定了视图布局并通过 URL 指向本地或远程数据文件。
- 除了声明性的 JSON 表示法之外,我们还开发了 Python、R 和 JavaScript API,以便用户能够以编程方式定义配置。
- 这些配置 API 支持在每种语言的原生面向对象范式中定义数据集、文件、视图、视图布局和视图协调。
JavaScript plugin API
JavaScript插件API
Para_01
- Vitessce JavaScript 包包含用于定义插件视图类型、协调类型、数据类型和文件类型的函数。
- 一旦定义了插件,就必须通过提供一个将在 Vitessce 配置中用于引用实现的名称来注册它们。
- 插件视图类型必须作为 React 组件来实现。
- 我们在 Vitessce 文档网站上提供了使用插件 API 的示例和教程。
Multimodal configuration
多模态配置
Para_01
- Vitessce 支持任意多模态数据集,采用单细胞生态系统中数据分析软件包使用的观察-特征矩阵约定,包括 MultiAssayExperiment、Seurat 和 MuData。
- 在这个模型中,观察对象是被测量的实体,例如细胞、分子、斑点、珠子或细胞核。
- 特征是关于实体所测量的特性,如基因、染色质可及性峰或表面蛋白。
- 特征值是所测量的数量,如表达水平、计数或强度。
- 可以在 Vitessce 配置中定义观察类型、特征类型和特征值类型的标识符,既用于数据也用于视图。
- 然后,Vitessce 根据观察类型、特征类型和/或特征值类型标识符将视图与数据匹配。
- 对于大多数视图和数据类型,这三种属性都会被使用,但根据特定视图的相关属性,可能会使用其中的一部分或全部。
- 例如,热图在加载数据时考虑所有三种属性,因为可视化包含特征、观察对象和值。
- 特定的热图可以通过这些属性唯一地识别。
- 相反,特征列表视图仅通过特征类型唯一识别,因此在加载视图的数据时不考虑观察类型和值类型。
Data organization
数据组织
Para_01
- 为了分离数据加载和渲染,并支持多模态实验,Vitessce 视图加载与数据集和数据类型相对应的数据,例如每个观测对象的空间坐标数组、每个观测对象的降维坐标、图像和观测对象-特征矩阵。
- 视图可以加载一个或多个数据集以及一个或多个数据类型对应的数据。
- 这些数据类型可能在某些轴上对齐(例如,以支持共享的观测对象或特征集合),或者不对齐(例如,以支持多个数据集的比较)。
Para_02
- 数据类型被独立加载,这样它们的数据可以包含在同一文件中或分布在独立的文件中,从而允许使用多种文件格式来加载每个数据集。
- Vitessce 定义了多种文件类型,对应于数据类型-文件格式对。
- 如果一种文件格式支持多种数据类型,则可以定义联合文件类型以简化配置(也就是说,允许为文件定义一次 URL 和文件选项,同时在内部作为多种数据类型暴露出来)。
- 例如,AnnData 对象可能包含多个观测值-特征矩阵、降维和空间坐标,可以使用特定于 AnnData 的高级联合文件类型进行配置。
- 然而,这些细节对于仅执行查找对应于各个数据类型的相应数据的视图实现来说是抽象化的。
On-demand loading of data subsets
按需加载数据子集
Para_01
- 为了扩展到大型数据集,在多种情况下延迟了数据加载:多尺度图像、基于多尺度位图的分割、基因组映射数据以及观察特征矩阵的每个特征子集。
- 高度多重化的多尺度(即金字塔)图像文件的加载是使用Viv实现的。如Manz等人所描述的11,Viv根据当前视口缩放级别(即分辨率)和目标位置(即X/Y),将图像加载为对应的数据瓦片,同时还有通道(C)、时间(T)和Z轴的选择。
- 在Vitessce中,这种方法不仅适用于主图像,还适用于用于细胞和细胞器分割的图像位图文件,这些文件也可以存储在多分辨率格式中。
Para_02
- Vitessce 在处理大型观测特征矩阵时利用了类似的数据分块方法。
- 这些矩阵可以存储在多种基于 Zarr 的文件格式中,特别是在使用 AnnData19,32、MuData21 和 SpatialData9 时。
- Zarr 支持分块(‘分块’)和压缩的多维数组,这些数组可以作为称为‘存储’的静态文件目录来提供。
- Moore 等人24进行的基准测试表明,访问 Zarr 数据至少与访问存储在层次化数据格式版本 5 (HDF5) 和 TIFF 中的相同数据一样快。
- 在高延迟云存储场景中,Zarr 的性能比 HDF5 高出一个数量级。
Para_03
- Zarr 块策略可以配置以优化特定用例。
- 在 Vitessce 中,当每个块存储许多观测值和很少的特征时,性能得到优化,这使得能够快速加载某一特定基因(即一个特征)在所有细胞(即所有观测值)中的表达值。
- 权衡的是,相同的块策略可能会导致在不同的用例下表现不佳,例如仅在一个细胞中加载所有基因的表达值。
- 我们还在补充图 7 中比较了 Zarr 和基于 CSV 的 AnnData 对象的完整大小。
Para_04
- 使用HiGlass可视化基因组浏览器轨道还会根据当前浏览器视口和缩放级别按需加载数据图块,如Kerpedjiev等人所述12。
- 在Vitessce中,我们将这种机制扩展到支持来自Zarr存储的基因组数据(除了HiGlass支持的现有文件格式),以消除对专用HiGlass服务器的依赖。这种扩展被实现为一个HiGlass插件数据获取器。
Coordinated multiple views
协调多个视角
Para_01
- Vitessce 采用信息可视化领域的协调多视图技术来实现比较任务,例如概览和细节、焦点与上下文、差异视图和小多重视图。
- 42,43 这些参考文献编号已被移除。
Coordination model
协调模型
Para_01
- Boukhelifa等人提出的协调模型被用来链接可视化、交互和数据属性的视图子集。
- 在这个模型中,视图不是直接相互连接,而是连接到称为协调范围的命名属性值。
- 视图可以连接的属性被称为协调类型。
- 因此,当视图针对给定的协调类型连接到相同的协调范围时,它们就被协调了。
JavaScript implementation
JavaScript 实现
Para_01
- Vitessce 是用 JavaScript 实现的一个可配置的 React 组件。
- 在根 React 组件之下,可视化视图和控制视图也实现了作为 React 组件。
- 可视化视图可以使用任何网络技术,包括 WebGL、SVG、CSS 和 HTML 画布元素。
- 主要的 Vitessce 组件被分发在一个 JavaScript 包中,该包还导出了一个面向对象的配置 API 和插件 API,用于定义自定义视图作为 React 组件、文件类型作为 JavaScript 类和协调类型。
Implementation of coordinated multiple views
协调多个视图的实现
Para_01
- 我们已在JavaScript中实现了上述协调模型,并使用JSON表示法将其整合到Vitessce配置模式中。
- 鉴于这可能对一般使用JavaScript实现协调的多个视图可视化有用,我们提供了一个独立的JavaScript包,该包可以与插件API一起使用。
Implementation of views
视图的实现
Para_01
- Vitessce 配置定义了一组包含交互式可视化的视图。
- 视图实现是相互独立的,并使用自定义的 React 钩子函数从协调模型中获取值并加载数据。
- 目前可用的视图包括散点图、空间图、热图、细胞集大小条形图、基因表达直方图、细胞集的基因表达小提琴图、细胞集管理器、基因选择、图像层控制器和细胞集的基因组浏览器。
- 可视化视图是使用自定义的 JavaScript 代码和现有的用于统计和地理空间数据可视化的 JavaScript 库实现的。
- 所使用的重大开源软件包列表和当前可用视图列表分别在补充表 1 和表 2 中提供。
Implementation of custom deck.gl layers
自定义deck.gl图层的实现
Para_01
- 这些视图的实现包括热力图、散点图和空间视图利用了基于deck.gl API的可扩展性来实现基于WebGL的数据可视化。
- Deck.gl不仅提供了用于渲染点、线、多边形和文本的高级JavaScript API("层"),而且还提供了开发自定义层的抽象,这些自定义层具有关联的自定义WebGL着色器程序。
- 热力图是通过使用一个自定义层实现的,该层在观察对象特征矩阵中对相邻值进行聚合计算,当多个值对应于屏幕上仅一个像素时。
- 这消除了热力图视图在低缩放级别下由于大型矩阵而可能出现的混叠和莫尔条纹现象,同时保持平滑的缩放和平移交互。
- 自定义热力图层还包含逻辑代码,限制平移交互仅在矩阵区域内进行,并根据缩放级别和文本长度确定如何显示轴刻度。
- 空间视图是通过多个自定义层实现的,其中包括一个从Viv图层扩展而来以渲染图像位图的图层。
- 空间视图和散点图视图都使用了一个自定义层,该层利用在WebGL中编写的定量颜色映射函数,在图形处理单元(GPU)上高效地将特征值映射到颜色。
- 这些功能使得在不同缩放级别下的视图能够平滑地呈现数据,同时保持良好的交互性和视觉效果。
Implementation of data loading
数据加载的实现
Para_01
- 数据是通过 HTTP 从本地或远程 web 服务器上的静态文件加载的。
- 配置文件中通过 URL 和文件类型的数组指定对应一个或多个数据集的文件。
- 某些文件类型接受选项,这些选项指定了内部文件组织的细节以启用查找(例如,通过指定相对路径来加载 Zarr 存储中的特定数组)。
- 每种文件类型由相应的 JavaScript 类加载,该类定义了一个必需的加载函数和可选的数据子集加载函数(在文件格式允许的情况下)。
- 数据加载类可以执行验证,特别是当使用诸如 JSON 这样的文件格式时,这些文件格式的内容可能差异很大。
- 支持的文件类型的列表见补充表 3。
Processing of data for use cases
用于案例的数据处理
Para_01
- 用于图2所示用例的数据处理使用了Python脚本、Jupyter笔记本和Snakemake管道(扩展数据图1、3和6)。
- Vitessce可视化配置使用了Vitessce Python包。
- 配置导出为JSON文件并上传到GitHub Gist服务以便通过URL引用。
- 从HuBMAP数据门户获得的文件由HuBMAP基础设施和参与协作组内开发的自动化管道进行处理。
Reporting summary
报告摘要
Para_01
- 关于研究设计的更多信息,请参阅本文链接的Nature Portfolio报告摘要。
- ,
Data Availability
Para_01
- 所有在图表中显示的数据都具有宽松的许可协议。
- smFISH 数据集可在 http://linnarssonlab.org/osmFISH/ 获得,参考文献为37。
- Visium 数据集可以从Scanpy Python包(v1.9.3)使用标识符‘V1_Human_Lymph_Node’获得。
- CODEX 数据集可以从HuBMAP数据门户使用标识符‘HBM287.WDKX.539’获得。
- CITE-seq 数据集可以从NCBI基因表达综合数据库使用标识符‘GSE100866’获得。
- Multiome 数据集可以从Mudatasets Python包(v0.0.2)使用标识符‘brain3k_multiome’获得,原始数据集可通过10x Genomics在 https://www.10xgenomics.com/resources/datasets/frozen-human-healthy-brain-tissue-3-k-1-standard-2-0-0 获得。
- 来自10x Genomics的Visium空间基因表达数据集是通过Scanpy数据集API获得的,原始数据集可以在 https://www.10xgenomics.com/resources/datasets/human-lymph-node-1-standard-1-1-0 获得。
- 三维多模态质谱成像数据集可以通过HuBMAP数据门户在 https://portal.hubmapconsortium.org/preview/multimodal-mass-spectrometry-imaging-data 获得。
- CODEX数据集可以通过HuBMAP数据门户在 https://doi.org/10.35079/HBM543.RSRV.265 和 https://portal.hubmapconsortium.org/browse/dataset/69d9c52bc9edb625b496cecb623ec081 获得。
- MALDI IMS(正模式)数据集可以通过HuBMAP数据门户在 https://doi.org/10.35079/hbm876.xnrh.336 和 https://portal.hubmapconsortium.org/browse/dataset/3ade70d66d10ed1d30fe005f672b2abf 获得。
Code availability
Para_01
- Vitessce 是一款免费的开源软件,受 MIT 许可证保护。
- Vitessce JavaScript 包的源代码可通过 GitHub 获取,网址为 https://github.com/vitessce/vitessce。
- JavaScript 包通过 NPM 分发,网址为 https://www.npmjs.com/package/vitessce。
- 包含配置 API 和 Jupyter 小部件的 Python 包的源代码可通过 GitHub 获取,网址为 https://github.com/vitessce/vitessce-python。
- Python 包通过 PyPI 分发,网址为 https://pypi.org/project/vitessce。
- 包含配置 API 和 htmlwidget 的 R 包可通过 GitHub 获取,网址为 https://github.com/vitessce/vitessceR。
- 文档和在线配置编辑器可在 http://vitessce.io 获取。
- 用于图中的 JSON 配置文件可通过 GitHub 获取,网址为 https://gist.github.com/keller-mark/17417ec5c17228b3ca78b5edd3a7b89e。
- 用于生成本文档中图中可视化效果的代码可通过 GitHub 获取,网址为 https://github.com/vitessce/paper-figures。
- 代码已存入 Zenodo,网址为 https://doi.org/10.5281/zenodo.11286222。
- 代码已存入 Zenodo,网址为 https://doi.org/10.5281/zenodo.11285945。
- 代码已存入 Zenodo,网址为 https://doi.org/10.5281/zenodo.11285962。
- 代码已存入 Zenodo,网址为 https://doi.org/10.5281/zenodo.11285991。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-02-19,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号