ChIP-Seq 分析流程-上游


写在前面的话: 参考使用的文件资料是由哈佛陈生物信息学核心 (HBC) 教学团队成员开发的。另外也看了多个公众号文章和书籍。 Website: https://hbctraining.github.io/Intro-to-ChIPseq/ Github: https://github.com/hbctraining/Intro-to-ChIPseq
碎碎念:因为会有代码存在,代码框中既有命令,又有注释和输出信息,为了避免误解,这里统一一下(仅限代码框内)。 “#” 字符后代表注释信息 “$” 字符后代表输入的命令代码,可主要关注这个。 其余信息均为命令输出内容
全流程概述
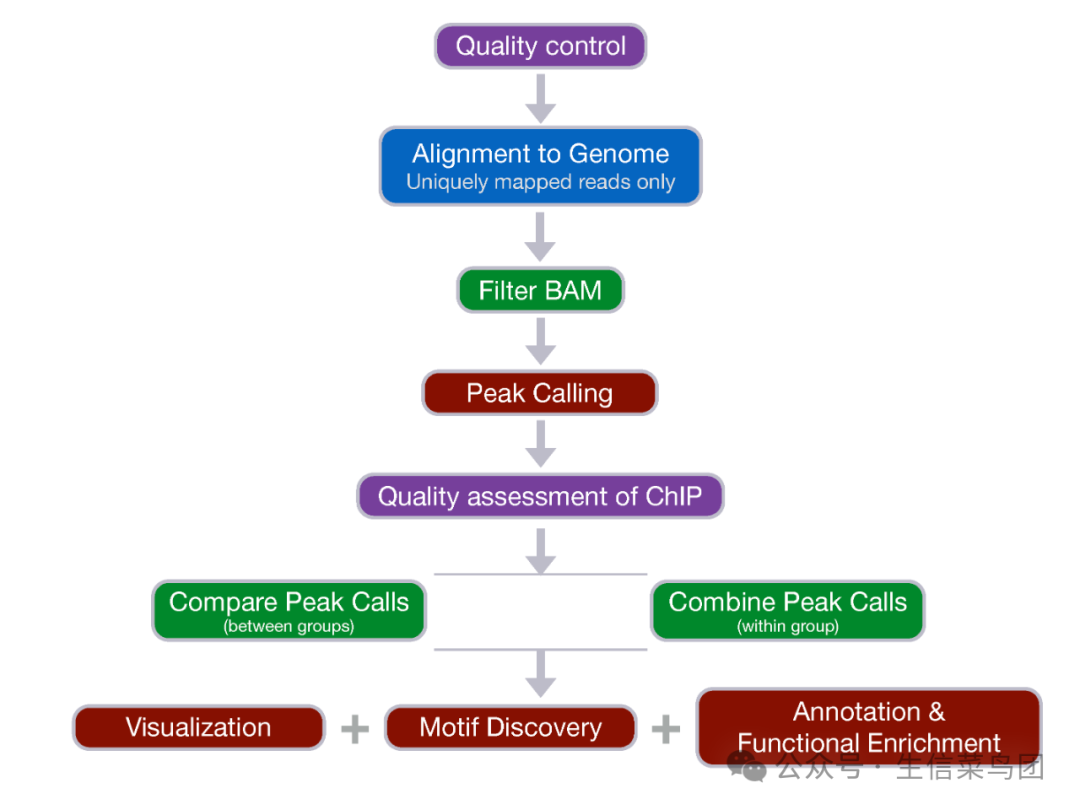
分析工具
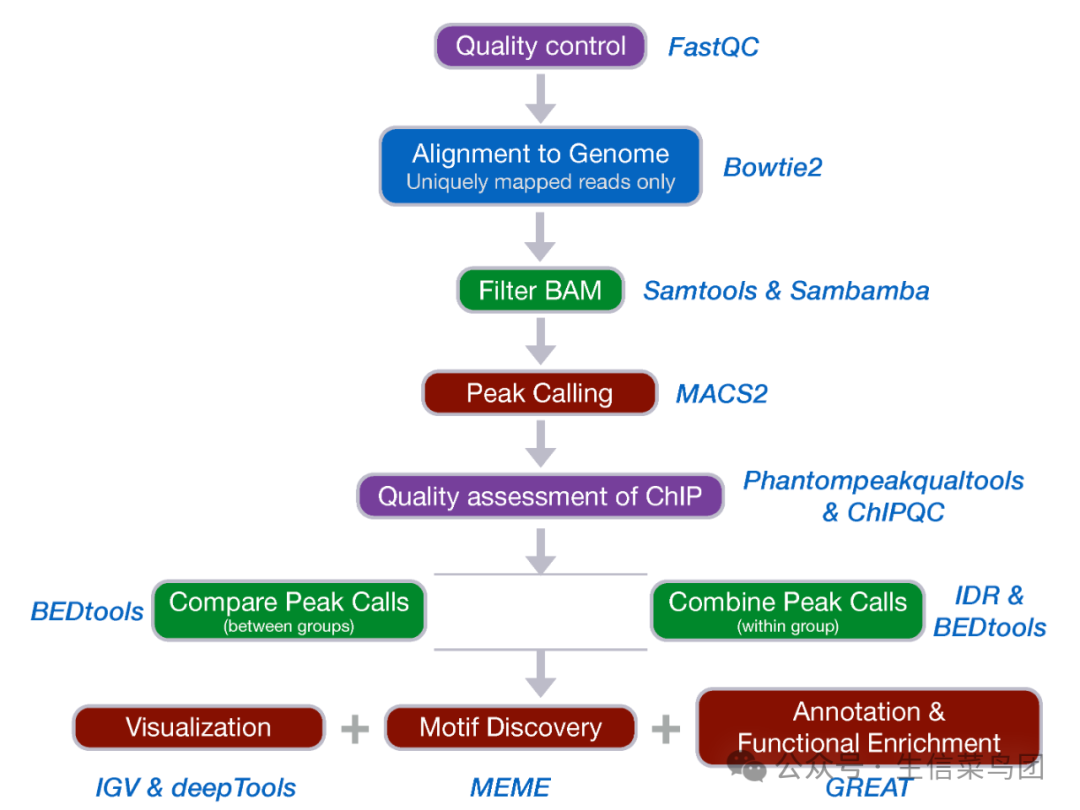
目前来说,分析工具肯定不止这些,每一步都有可替换的工具,甚至有些工具已经有所更新。但我感觉都差不多,感觉不行咱就换!
数据格式
实际上,生信分析只是分析数据。软件工具只是将数据从一种格式转换为另一种格式。新生成的数据通常会有不同的优化。它可能更加丰富,因为它可能结合了来自其他数据的信息。但它仍然是数据——生物学洞察力来自于对这些数据的理解和研究。
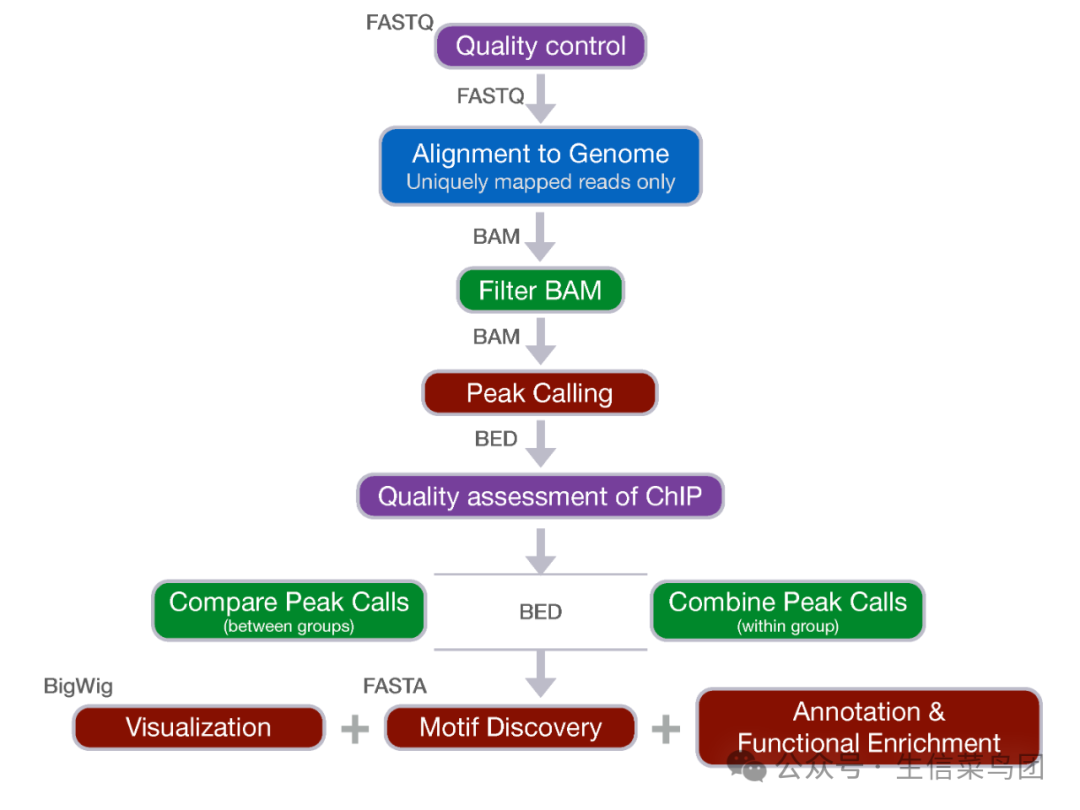
分析过程
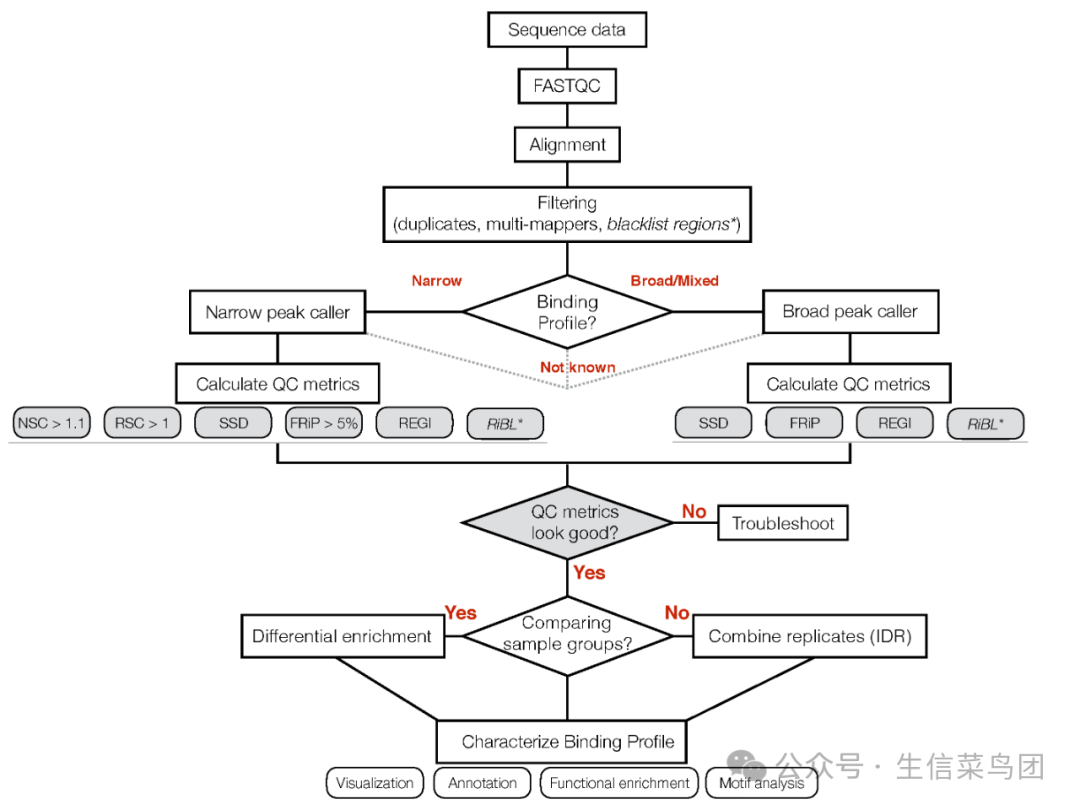
创建目录
让我们直接开始吧!
这里使用的是学习资料给到的目录结构,但这个结构好坏因人而异,这里为了方便,我也就全盘接收了。
- 创建项目名称
$ mkdir chipseq
$ cd chipseq/
- 创建分析目录
$ mkdir raw_data reference_data scripts logs meta
$ mkdir -p results/fastqc results/bowtie2
- 查看目录结构
# 使用tree 查看目录结构
$ tree
.
|-- logs
|-- meta
|-- raw_data
|-- reference_data
|-- results
| |-- bowtie2
| `-- fastqc
`-- scripts
logs:跟踪运行的命令和使用的具体参数,同时还记录运行命令时生成的任何标准输出。meta:用于描述您正在使用的样本的任何信息,我们将其称为元数据raw_data:用于计算分析之前获得的任何未修改的(原始)数据,例如来自测序中心的 FASTQ 文件。我们强烈建议在分析过程中不要修改此目录。reference_data:用于分析的参考基因组相关的已知信息,例cd如基因组序列(FASTA)、与基因组相关的基因注释文件(GTF)。results:用于您在工作流程中实施的不同工具的输出。在此文件夹中创建特定于工作流程的每个工具/步骤的子文件夹。scripts:用于您编写并用于运行分析/工作流程的脚本。
下载原始数据
原始数据依旧是 FASTQ 格式。
此资料使用的数据包含六个样本。其中两个样本作为 control.
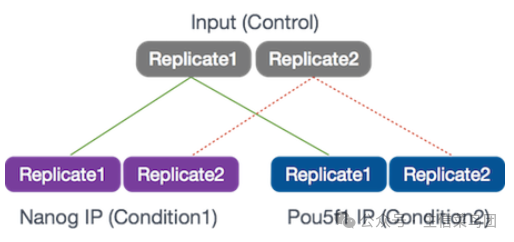
这里简单解释一下样本情况:这个测序的目的是找到 Nanog 和 Pou5f1 两个转录因子的结合位点。在实验部分,首先将细胞分为三份(不一定等分),分别不做抗体富集(input control)、Nanog 抗体富集和 Pou5f1 抗体富集,生成三组数据,再进行重复,生成六个测序样本。
原始数据链接
原教程的原始数据比较难找,就不详细说明过程了,这里直接给出下载 URL 好了。
- Nanog
# rep1
https://www.encodeproject.org/files/ENCFF000OQA/@@download/ENCFF000OQA.fastq.gz
# rep2
https://www.encodeproject.org/files/ENCFF000OQM/@@download/ENCFF000OQM.fastq.gz
- Pou5f1
# rep1
https://www.encodeproject.org/files/ENCFF000ORV/@@download/ENCFF000ORV.fastq.gz
# rep2
https://www.encodeproject.org/files/ENCFF000ORW/@@download/ENCFF000ORW.fastq.gz
- Input
# rep1
https://www.encodeproject.org/files/ENCFF000OSS/@@download/ENCFF000OSS.fastq.gz
# rep2
https://www.encodeproject.org/files/ENCFF000OST/@@download/ENCFF000OST.fastq.gz
批量下载
现在创建一个批量下载脚本
$ cat down_fq.sh
#!/bin/bash
URL=~/project/chipseq/raw_data/url.txt
dir=~/project/chipseq/raw_data
cat ${URL} | parallel --dry-run aria2c -x 5 -d ${dir} {}
cat ${URL} | parallel aria2c -x 5 -d ${dir} {}下载后解压
$ gunzip *.gz
查看原始数据情况,我这里推荐使用 seqkit 工具去统计一下我们的原始数据情况,可以比对下看是否有缺失。
$ ls *fastq
ENCFF000OQA.fastq ENCFF000OQM.fastq ENCFF000ORV.fastq ENCFF000ORW.fastq ENCFF000OSS.fastq ENCFF000OST.fastq
$ seqkit stats *fastq
processed files: 6 / 6 [======================================] ETA: 0s. done
file format type num_seqs sum_len min_len avg_len max_len
ENCFF000OQA.fastq FASTQ DNA 15,329,142 551,849,112 36 36 36
ENCFF000OQM.fastq FASTQ DNA 63,707,371 2,293,465,356 36 36 36
ENCFF000ORV.fastq FASTQ DNA 18,153,153 653,513,508 36 36 36
ENCFF000ORW.fastq FASTQ DNA 35,253,613 1,269,130,068 36 36 36
ENCFF000OSS.fastq FASTQ DNA 55,951,481 2,014,253,316 36 36 36
ENCFF000OST.fastq FASTQ DNA 27,548,706 1,377,435,300 50 50 50
为了便于查看,我们可以手动改个名字,我根据需要将六个样本改成了这样。
$ ls *fastq
H1hesc_Input_Rep1.fastq H1hesc_Input_Rep2.fastq H1hesc_Nanog_Rep1.fastq H1hesc_Nanog_Rep2.fastq H1hesc_Pou5f1_Rep1.fastq H1hesc_Pou5f1_Rep2.fastq
下载参考基因组
$ cd reference_data/
# 下载fa文件
$ aria2c -x 5 -d ./ https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/001/405/GCF_000001405.40_GRCh38.p14/GCF_000001405.40_GRCh38.p14_genomic.fna.gz
# 下载gtf文件
$ aria2c -x 5 -d ./
https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/001/405/GCF_000001405.40_GRCh38.p14/GCF_000001405.40_GRCh38.p14_genomic.gtf.gz
解压
$ gunzip *.gz
质量评估
$ cd raw_data
$ fastqc *.fastq
所有数据下载完后就可以正式进行数据分析了。简单进行数据质量评估。
质评结果就不详细说明了,因为现在测序技术越来越成熟,很多数据本来质量就很好,就不需要过滤了,如果查看评估结果后觉得需要过滤,就使用一些常见的过滤软件默认参数过滤一下就好了,基本上问题不大。
Bowtie 比对
Bowtie2是一种快速准确的对齐工具,它使用基于 Burrows-Wheeler 变换方法的 FM 索引对基因组进行索引,以保持对齐过程的内存要求较低。Bowtie2支持间隙、局部和双端对齐模式,最适合至少 50 bp 的读取(较短的读取长度应使用 Bowtie1)。默认情况下, Bowtie2将执行全局端到端读取对齐,这最适合质量修剪的读取。但是,它还有一种局部对齐模式,它将执行软剪辑以从未修剪的读取中去除质量较差的碱基或适配器。我们将使用此选项,因为我们没有修剪我们的 reads。
构建索引
如果没有构建索引,第一步需要构建索引。
$ cd reference_data
$ bowtie2-build GCF_000001405.40_GRCh38.p14_genomic.fna GCF_000001405.40_GRCh38.p14_genomic
比对
我们先对单个原始 fastq 文件进行处理
$ mkdir results/bowtie2
$ bowtie2 -p 4 -q --local \
-x reference_data/GCF_000001405.40_GRCh38.p14_genomic \
-U raw_data/H1hesc_Input_Rep1.fastq \
-S results/bowtie2/H1hesc_Input_Rep1_aln_unsorted.sam
过滤reads
ChIP-seq数据中包含多重映射的读取(reads映射到参考基因组的多个位置)。允许多重映射的读取可以增加可用读取的数量和峰值检测的灵敏度;然而,假阳性的数量也可能增加。因此,我们需要过滤我们的对齐文件,只包含唯一映射的读取,以提高位点发现的置信度和改善可重复性。由于Bowtie2中没有参数可以只保留唯一映射的读取,我们需要执行以下步骤来生成只包含唯一映射读取的对齐文件:
- 将对齐文件格式从SAM转换为BAM
- 按读取坐标位置对BAM文件进行排序
- 过滤以仅保留唯一映射的读取(这也将删除任何未映射的读取)
代码如下:
- SAM 转 BAM
$ samtools view -h -S -b \
-o H1hesc_Input_Rep1_aln_unsorted.bam \
H1hesc_Input_Rep1_aln_unsorted.sam
- BAM 排序
$ sambamba sort -t 2 \
-o H1hesc_Input_Rep1_aln_sorted.bam \
H1hesc_Input_Rep1_aln_unsorted.bam
- 筛选唯一比对
$ sambamba view -h -t 2 -f bam \
-F "[XS] == null and not unmapped and not duplicate" \
H1hesc_Input_Rep1_chr12_aln_sorted.bam > H1hesc_Input_Rep1_chr12_aln.bam
自动化比对&过滤
正常来说,我们有六个样本,上述比对和过滤步骤我们要重复六次,这里我们使用一个自定义脚本可以更快的完成这个过程。
首先我们先产生样本 ID 文件
$ ls *fastq | xargs -n 1 -I {} basename {} .fastq > ID.txt
$ cat ID.txt
H1hesc_Input_Rep1
H1hesc_Input_Rep2
H1hesc_Nanog_Rep1
H1hesc_Nanog_Rep2
H1hesc_Pou5f1_Rep1
H1hesc_Pou5f1_Rep2
创建脚本
#!/bin/bash
genome=~/project/chipseq/reference_data/GCF_000001405.40_GRCh38.p14_genomic
ID=~/project/chipseq/raw_data/ID.txt
align=~/project/chipseq/results/bowtie2
# Run bowtie2
cat ${ID} | parallel \
bowtie2 -p 6 -q --local -x ${genome} -U {}.fastq -S ${align}/{}_unsorted.sam
# Create BAM from SAM
cat ${ID} | parallel \
samtools view -h -S -b -@ 6 -o ${align}/{}_unsorted.bam ${align}/{}_unsorted.sam
# Sort BAM file by genomic coordinates
cat ${ID} | parallel \
sambamba sort -t 6 -o ${align}/{}_sorted.bam ${align}/{}_unsorted.bam
# Filter out duplicates
cat ${ID} | parallel \
sambamba view -h -t 6 -f bam -F "[XS] == null and not unmapped " ${align}/{}_sorted.bam > ${align}/{}_aln.bam
# Create indices for all the bam files for visualization and QC
cat ${ID} | parallel \
samtools index ${align}/{}_aln.bam
运行脚本
bash align_filter.sh
Peak calling
这一步就是找到我们的转录因子结合的区域了,也就是 peak 区域。
我们使用 macs2 进行 peak calling
$ mkdir -p ~/chipseq/results/macs2
$ macs2 callpeak -t bowtie2/H1hesc_Nanog_Rep2_aln.bam -c bowtie2/H1hesc_Input_Rep2_aln.bam -f BAM -g 1.3e+8 --outdir macs2 -n Nanog-rep2 2> macs2/Nanog-rep2-macs2.log
$ macs2 callpeak -t bowtie2/H1hesc_Pou5f1_Rep1_aln.bam -c bowtie2/H1hesc_Input_Rep1_aln.bam -f BAM -g 1.3e+8 --outdir macs2 -n Pou5f1-rep1 2> macs2/Pou5f1-rep1-macs2.log
$ macs2 callpeak -t bowtie2/H1hesc_Pou5f1_Rep2_aln.bam -c bowtie2/H1hesc_Input_Rep2_aln.bam -f BAM -g 1.3e+8 --outdir macs2 -n Pou5f1-rep2 2> macs2/Pou5f1-rep2-macs2.log
到这里上游分析已经结束了,下游分析和每一步的详细讨论后续会继续更新~~~。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-02-10,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号