细胞死亡途径指南 | Nature Reviews Molecular Cell Biology

细胞死亡途径指南 | Nature Reviews Molecular Cell Biology

生信菜鸟团
发布于 2024-12-27 17:11:34
发布于 2024-12-27 17:11:34

Basic Information
- 英文标题: A guide to cell death pathways
- 中文标题:细胞死亡途径指南
- 发表日期:18 December 2023
- 文章类型:Review Article
- 所属期刊:Nature Reviews Molecular Cell Biology
- 文章作者:Junying Yuan | Dimitry Ofengeim
- 文章链接:https://www.nature.com/articles/s41580-023-00689-6
Abstract
Para_01
- 由专门的分子机器介导的调节性细胞死亡,即程序性细胞死亡,在健康和疾病中起着重要的作用。
- 凋亡、坏死性凋亡和焦亡是三种这样的程序性细胞死亡方式。
- 半胱氨酸蛋白酶家族的caspase作为程序性细胞死亡的关键调节因子。
- 在凋亡过程中,caspase的级联激活介导信号传导和细胞破坏,而当激活的caspase切割gasdermins时,就会发生焦亡,后者可以在质膜上形成孔隙。
- 坏死性凋亡是一种由RIPK3和MLKL介导的、与caspase无关的程序性坏死形式,它被caspase-8介导的RIPK1切割所抑制。
- 细胞生存所必需的细胞内环境稳定机制的破坏,如正常的离子和氧化还原平衡以及溶酶体流量,也可以在不触发程序性细胞死亡机制的情况下诱导细胞死亡。
- 兴奋性毒性、铁死亡和溶酶体细胞死亡就是这样的细胞死亡方式的例子。
- 在这篇综述中,我们提供了主要的细胞死亡机制的概述,强调了它们复杂的调控和执行最新见解,以及它们与人类疾病的相关性。
Introduction
Para_01
- 细胞死亡是生物体发育和成年稳态的基本组成部分。
- 在发育过程中消除多余的细胞对于确保正常的形态发生和器官发生是重要的。
- 在成年生活中,消除自反应免疫细胞、癌细胞和受损细胞对于维持稳态是必要的。
- 细胞死亡过多或过少都可能导致人类疾病——神经退行性疾病涉及本应长寿以维持神经功能的神经元的死亡,而癌症的一个标志就是无法消除携带癌变突变的细胞。
Para_02
- 可以根据形态学将不同类型的细胞死亡进行分类。
- 例如,凋亡和坏死就是根据它们独特的形态学特征来描述的。
- 凋亡的特点是细胞质和核的浓缩,但不损害细胞质膜的完整性。
- 相比之下,早期细胞膜完整性的丧失和由于离子失衡导致的细胞质室扩张是坏死的典型特征。
- 在1960年代,Richard Lockshin和Carroll M. Williams提出了'程序性细胞死亡'这个术语,用以描述在蚕蛾蜕皮过程中间隔肌的退化,这暗示了肌肉退化的遗传调控。
- 但是,对于细胞死亡分子机制的了解要晚得多。
- 过去三十年的研究揭示了介导凋亡、坏死性凋亡和焦亡的特定分子机器,并识别出编码其组件的哺乳动物基因。
- 我们建议将凋亡、坏死性凋亡和焦亡统称为'程序性细胞死亡'。
Para_03
- 除了由于程序性细胞死亡途径的激活导致的细胞死亡——如凋亡、坏死和焦亡——细胞还可能在维持正常细胞稳态所必需的关键细胞生存机制被失活或破坏时死亡。
- 例如,这包括细胞质和/或细胞内膜完整性的丧失、错误折叠蛋白的积累、兴奋性毒性、氧化应激和脂质过氧化。
- 兴奋性毒性是由于特定氨基酸(如谷氨酸)的毒性积累所致,主要发生在神经元中。
- 铁死亡是由于细胞对抗脂质过氧化的抗氧化机制失活所致。
- 病理性的溶酶体酸性水解酶释放,其正常功能是分解大分子以供细胞回收,可能导致溶酶体细胞死亡。
- 细胞也可能被另一个细胞在一种称为侵入的过程中被活活吞噬。
Para_04
- 随着人们越来越认识到失调的细胞死亡过程在人类疾病中起着重要作用,人们正在投入大量精力去识别涉及特定人类病理状况的细胞死亡机制——无论是细胞死亡是由程序性细胞死亡机制的激活、细胞存活机制的丧失还是二者的结合所致。
- 在本综述中,我们首先概述了程序性细胞死亡的触发因素、机制和调节,包括凋亡、焦亡和坏死性凋亡。
- 然后,我们描述了吞噬、兴奋性毒性、铁死亡、溶酶体细胞死亡和有丝分裂灾难的机制,这些都是通过破坏不同的细胞稳态促存活机制介导的。
- 我们还讨论了由于破坏细胞存活和程序性细胞死亡而导致的这些细胞死亡方式之间的联系。
- 最后,我们简要讨论了细胞死亡在人类疾病中的最新认识。
Apoptosis
Para_01
- 细胞死亡的中介机制最早是在秀丽隐杆线虫中描述的,后来在哺乳动物中发现了秀丽隐杆线虫细胞死亡效应蛋白的同源物,包括半胱氨酸天冬氨酸蛋白酶(caspases)和BCL-2蛋白(见表1)。
- 哺乳动物细胞的凋亡可以分为内在凋亡和外在凋亡,这两者在细胞对凋亡触发信号的感应和整合方式上有所不同。
- 外在凋亡是通过激活位于质膜上的死亡受体来介导的,而内在凋亡的启动则不涉及死亡受体的参与(见图1)。
Fig. 1: Intrinsic and extrinsic apoptosis.
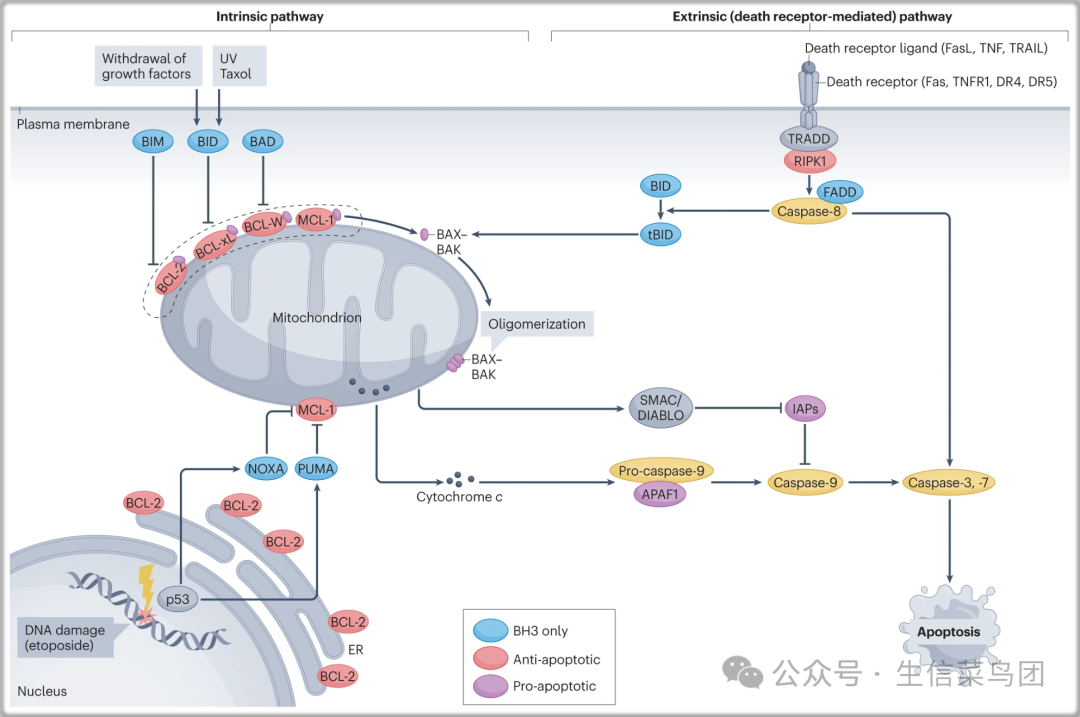
- 外在凋亡(右)是由位于质膜上的死亡受体(如 TNFR1、Fas 和 TRAIL 受体 DR4 和 DR5)通过其特定配体(分别是 TNF、FasL 和 TRAIL)激活介导的。内在凋亡(左)可以通过生长因子撤除、线粒体损伤、DNA 损伤和化疗药物如紫杉醇激活。BCL-2 家族中仅含 BH3 结构域的成员激活,如 p53 诱导的 NOXA 和 PUMA 的转录,BAD 和 BIM 的翻译后修饰,以及 BID 被 caspase-8 裂解,通过失活抗凋亡的 BCL-2 家族成员如 BCL-2、BCL-xL 和 MCL-1,以及激活促凋亡的 BCL-2 家族成员 BAX 和 BAK 的寡聚化,导致线粒体损伤。
- 线粒体损伤导致细胞色素 c 和第二个线粒体衍生的胱天蛋白酶激活剂(SMAC;也称为 DIABLO)的释放,以促进 APAF1 介导的胱天蛋白酶-9 的激活。激活的胱天蛋白酶-9 反过来裂解下游的胱天蛋白酶,胱天蛋白酶-3 和胱天蛋白酶-7,以介导内在凋亡的执行。激活的胱天蛋白酶-3 和胱天蛋白酶-7 也可以进行反馈激活上游胱天蛋白酶,以允许胱天蛋白酶级联的放大。
- 内质网;紫外线。
Intrinsic apoptosis
内在凋亡
Para_01
- 内源性凋亡可以在细胞发生诸如DNA损伤、生长因子撤除和线粒体损伤等改变后被激活(图1)。
- 锚定丢失诱导的细胞凋亡(anoikis)也是一种内源性凋亡形式,可以在某些细胞失去整合素介导的与细胞外基质的连接时被激活。
Para_02
- BCL-2蛋白家族是内在凋亡途径的关键上游调节因子。
- BCL-2,作为秀丽隐杆线虫CED-9的哺乳动物同源物,最初是在人类滤泡性淋巴瘤中作为一种癌基因被分离出来的,这种淋巴瘤具有t(14;18)转位,并且促进细胞存活而不是致癌细胞增殖。
- 哺乳动物的BCL-2家族包括抗凋亡和促凋亡成员,类似于秀丽隐杆线虫中的CED-9和EGL-1。
- BCL-2蛋白家族的特征是包含一个到四个BCL-2同源(BH)结构域,这个家族既包括抗凋亡成员也包括促凋亡成员。
- 抗凋亡BCL-2蛋白的高水平表达会抑制这一细胞死亡途径的激活。
- 凋亡在动物发育和成年生活中至关重要,因为抗凋亡蛋白BCL-2、BCL-xL或MCL-1的遗传消融可能导致小鼠死亡或产生有害表型。
- BCL-2家族中的BH结构域介导促凋亡和抗凋亡家族成员之间的相互作用,以抑制或激活凋亡。
- 在DNA损伤、氧化应激或营养剥夺的响应中,促凋亡BCL-2家族的转录和翻译后激活对传递促凋亡信号以驱动线粒体外膜透化至关重要。
- 在这个过程中,仅含BH3的促凋亡BCL-2蛋白,如BIM、BID、BAD、NOXA和PUMA,利用它们的BH3结构域既主动抑制抗凋亡BCL-2蛋白,如BCL-2、BCL-xL、MCL-1和BCL-W,也直接激活促凋亡BAX和BAK,驱动线粒体外膜形成寡聚孔。
- 线粒体外膜的破裂导致细胞色素c和第二个线粒体来源的caspase激活剂(SMAC,也称为DIABLO)的释放,从而促进下游caspase的激活。
- APAF1,作为CED-4的哺乳动物同源物和NOD家族的一员,形成七聚体凋亡小体以促进caspase-9的激活,进而介导其他下游caspase,如caspase-3和caspase-7的裂解,这些caspase可以裂解数百个蛋白底物以促进细胞破坏。
- 然而,简单的上下游caspase级联观念应该修正,因为小鼠中caspase-3和caspase-7的双重敲除也可以延迟线粒体损伤和细胞死亡。
- 因此,凋亡可能被视为一个具有自我放大能力的循环级联。
Para_03
- 由离子辐射或化疗药物(如依托泊苷和紫杉醇)引起的DNA损伤激活内源性凋亡,构成了传统癌症放疗和化疗的基础。
- 然而,过去十年的研究表明,哺乳动物细胞死亡机制存在冗余性。
- 在Bax–/–Bak1–/–细胞或Bax–/–Bak1–/–双重敲除小鼠中同时敲除Bax和Bak1基因会导致对凋亡的高度抵抗,但仍有少数突变小鼠可以出生并生存数月。
- 这表明,当在发育过程中凋亡失败时,Bax–/–Bak1–/–细胞可能激活其他细胞死亡机制和/或选择其他机制以消除不需要的细胞。
- 此外,尽管DNA损伤引起的细胞死亡可以通过促进线粒体损伤和下游caspase活化来激活凋亡,但DNA损伤诱导的凋亡可能仅构成细胞死亡反应的一部分,因为严重的DNA损伤可能导致细胞通过有丝分裂灾难死亡(见下文)。
- 因此,当程序性细胞死亡机制被阻断时,破坏细胞稳态的促生存机制可能有助于哺乳动物细胞消除。
Extrinsic apoptosis
外在性凋亡
Para_01
- 外在性凋亡是由细胞质膜定位的死亡受体的激活介导的,这些死亡受体特征是存在一个被称为死亡域(DD)的细胞内蛋白质-蛋白质相互作用域,例如Fas(也称为CD95)、TNFR1、TRAIL(DR4)和TRAIL-R2(DR5),通过与它们的同源配体相互作用。
- 这些通路与人类疾病相关,因为Fas及其配体FasL的遗传缺陷会导致一种罕见的人类自身免疫状况,称为自身免疫性淋巴增生综合征,这种病症可以通过Fas和FasL突变小鼠进行表型复制。
- 活化的T细胞表达的FasL通过促进适配蛋白FADD和caspase-8的招募来诱导凋亡,这是通过与目标细胞上的Fas的DD相互作用实现的。
- 自身免疫性淋巴增生综合征相关的Fas DD中的突变导致FADD的招募丧失和caspase-8激活的抑制。
- 这些发现强调了caspase-8激活在Fas介导的凋亡中的重要性。
Fig. 2: Necroptosis mediated by DD-containing receptors and pattern-recognition receptors.
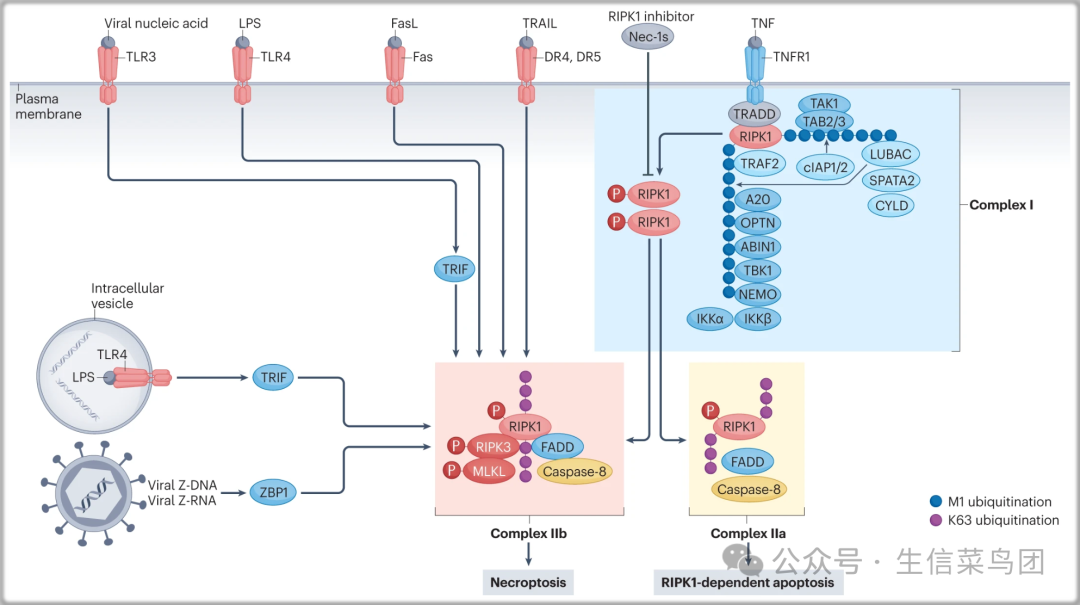
- 在TNF激活下,三聚化的TNFR1的细胞质死亡域(DD)介导形成一个瞬时的细胞内复合物,称为复合物I,该复合物招募适配蛋白TRADD、含DD的蛋白激酶RIPK1以及几个E3泛素连接酶,包括TRAF2、cIAP1、cIAP2、线性泛素链组装复合物(LUBAC)、泛素编辑酶A20和去泛素化酶圆柱状病态复合物(CYLD和SPATA2)以及多个泛素结合蛋白,包括NEMO、ABIN1和OPTN。
- 在复合物I中,RIPK1通过Lys63连接和线性Met1连接的泛素链被快速多泛素化,这介导了TAK1、TBK1和IKK复合物的招募和激活。
- 当活细胞中的RIPK1激酶未被激活时,IκB的磷酸化及其后续的泛素-蛋白酶体系统介导的降解导致核内NF-κB的激活(未显示)。
- 复合物I的失调促进RIPK1的激活(由pS166 RIPK1标记),这导致形成两个替代的细胞质复合物,复合物IIa或复合物IIb,分别介导RIPK1依赖的凋亡和坏死。
- 复合物IIa包括适配器FADD蛋白、caspase-8和RIPK1,促进caspase-8的激活,进而裂解下游的caspase如caspase-3,最终导致凋亡。
- 当caspase-8的激活被抑制时,活化的RIPK1激酶与RIPK3结合形成复合物IIb。
- 活化的RIPK3进而磷酸化MLKL,通过破坏质膜完整性介导坏死的执行。
- 当Toll样受体(TLR3)和TLR4分别被其特异配体病毒核酸和细菌脂多糖(LPS)激活时,在缺乏caspase功能的细胞中,可促进TIR结构域包含的适配器诱导IFNβ(TRIF)与RIPK3结合介导坏死。
- 当病毒Z-DNA或Z-RNA激活时,Z-DNA结合蛋白1(ZBP1;也称为DAI)通过其各自的RHIMs(受体相互作用蛋白同型交互基序)与RIPK3结合,也可以在缺乏caspase功能的情况下促进坏死。
- DR4或DR5被TRAIL激活、Fas被FasL激活、TLR4被LPS激活也已被证明在某些条件下可以促进坏死。
Para_02
- 另一个具有重大医学意义的死亡受体介导的外源性凋亡的例子是TNF介导的TNFR1激活。TNF激活TNFR1会导致迅速形成一个短暂的细胞内信号复合物,称为复合物I,它在做出细胞生死决定中起着关键作用。
- 激活的TNFR1的细胞内DD结构域招募含有DD结构域的适配蛋白TRADD和RIPK1,这是一种含有DD结构域的激酶。
- TRADD转而介导招募TRAF2、TRAF5、cIAP1和cIAP2,它们在复合物I上催化K63连接、K48连接和K11连接的多聚泛素化。
- 由cIAP1和cIAP2产生的多聚泛素链用于招募其他复合物I组分,如线性泛素链组装复合物(LUBAC)。
- LUBAC包括催化组分HOIP(RNF31)、HOIL1和SHARPIN,它们在复合物I的组分如RIPK1、NEMO、A20、TRADD和TNFR1上催化M1连接的泛素化。
- 复合物I上的泛素链还用于招募关键的激酶,包括TAK1–TAB1–TAB2–TAB3复合物和NEMO–IKKα–IKKβ激酶复合物,以激活NF-κB,并对RIPK1进行抑制性磷酸化。
- 在活细胞中,正确组装和泛素化的复合物I对于经典NF-κB的激活至关重要,NF-κB是一种促生存和促炎性的转录途径,控制CFLAR和TNFAIP3的表达,这两个基因编码凋亡和炎症的关键调节因子。
- A20,由TNFAIP3编码,是一种泛素链编辑酶,它改变RIPK1的泛素化模式以控制其激酶活性。
- CFLAR编码多个c-FLIP异构体,其氨基酸序列与caspase-8相似,但缺乏酶活性,可以直接与caspase-8结合并调节其激活。
Para_03
- TNFR1被TNF激活后,可能促进RIPK1独立的凋亡或RIPK1依赖的凋亡(RDA)。
- 使用TNF和环己酰亚胺(后者阻断NF-κB通路的下游翻译)的处理可以诱导RIPK1独立的凋亡,而RIPK1泛素化和磷酸化的失调可以促进RDA。
- cIAP1、cIAP2、LUBAC或NEMO的缺失,TAK1、TBK1或IKKs的抑制,或是在RIPK1中突变一个泛素接受位点(K377R),会导致RIPK1激酶活性的过度激活。
- 激活的RIPK1介导形成一个细胞质复合体,称为复合体IIa,包含RIPK1、FADD和caspase-8,以介导caspase-8的激活,从而促进RDA。
- 因此,NF-κB激活的关键介质,包括TAK1、IKKs、TBK1、NEMO、cIAP1、cIAP2和LUBAC,在抑制和调节RDA中也扮演着重要角色。
- RIPK1的激活作为形成复合体IIa的关键检查点,而抑制RIPK1的激酶活性在阻断RDA中非常有效。
Necroptosis
Para_01
- 坏死传统上被认为是细胞死亡的被动形式,因此被认为是无调控的。
- 自2000年代中期以来的突破性发现,识别了RIPK1、RIPK3和MLKL,这些基因编码在哺乳动物和高等脊椎动物的基因组中,介导了一种被称为坏死性程序性细胞死亡途径的坏死,即坏死性凋亡。
- 坏死性凋亡代表了发现的第一个介导坏死的程序性细胞死亡机制。
- 在凋亡缺陷条件下,死亡受体包括TNFR1、Fas、DR4和DR5,被其相应的配体激活,可以促进坏死性凋亡。
- 坏死性凋亡首次被定义为在用坏死素-1(Nec-1)处理后对caspase独立的坏死具有抑制作用。
- Nec-1及其改进的小分子类似物,如Nec-1s,后来被发现是RIPK1激酶活性的抑制剂。
- RIPK1的激活可以在凋亡功能正常条件下介导RDA,在凋亡缺陷条件下介导坏死性凋亡。
- 因此,RIPK1激活在介导TNFR1下游的多重有害反应中扮演着重要角色,这些反应取决于遗传背景、细胞类型和疾病条件。
Para_02
- caspase 8介导的RIPK1中間域(在鼠RIPK1的D325之后和在人RIPK1的D324之后)的裂解抑制了RIPK1激酶活性的激活,这是通过将N端激酶域与中間域和C端DD分开来实现的,后者介导RIPK1的二聚化以促进其激酶激活。
- 在RIPK1中罕见的有害错义突变,这些突变改变了caspase 8的裂解位点,定义了人类早期发病的自炎性疾病的一个类别,这类疾病的特点是周期性发热。
Para_03
- 在caspase缺陷细胞中激活RIPK1会促进复合物IIb(坏死小体)的形成,该复合物包括RIPK3和MLKL,介导坏死性凋亡
- 在坏死性凋亡过程中,活化的RIPK1与RIPK3之间的相互作用是由它们各自的同型RIP同型相互作用基序(RHIM)介导的,这些基序形成一个RHIM介导的类淀粉样结构,促进RIPK3的激活
- 活化的RIPK3,在小鼠RIPK3中标记为Thr231和Ser232的磷酸化,在人类RIPK3中标记为Ser227的磷酸化,转而介导伪激酶MLKL在人类MLKL中的Ser358或在小鼠MLKL中的Ser345的磷酸化
- 这种磷酸化促进MLKL的寡聚化,以及其N端四螺旋束(4HB)结构域中的带电氨基酸与质膜中的磷脂酰肌醇磷酸的相互作用
- 结果,寡聚化的MLKL嵌入质膜并形成孔洞,导致坏死
- 多个Ripk1激酶死亡敲入突变小鼠系并未表现出发育缺陷,成年表型正常,但对抗肿瘤坏死因子注射具有高度抵抗力,这种注射能在野生型小鼠中迅速导致死亡
- Ripk3基因敲除小鼠和Mlkl基因敲除小鼠也表现出对肿瘤坏死因子注射的抵抗性
- 这些结果表明坏死性凋亡在介导肿瘤坏死因子引起的败血症中的重要性
Para_04
- 与Fas和TNFR1相似,DD受体DR4和DR5在缺乏caspase活性的细胞中,可以被其特异配体TRAIL激活,进而引发坏死85,86。
- 此外,在缺乏caspase活性的细胞中,坏死还可以由模式识别受体(PRRs),例如Toll样受体3(TLR3)、TLR4和Z-DNA结合蛋白1(ZBP1)的激活引起87,88,89,90(图2)。
- 由TLR3和TLR4介导的坏死是通过TIR结构域包含的适配器诱导IFNβ(TRIF,也称为TICAM1)通过RHIM依赖的方式与RIPK3结合来驱动的。
- ZBP1通过病毒Z-RNA或Z-DNA与干扰素刺激的激活可以促进坏死,作为宿主防御反应的一部分,独立于RIPK1和TNFR188,89。
- ZBP1是细胞质核酸的感应器,并且可以通过与RIPK3的两个RHIM结构域结合形成坏死体来与病毒Z-DNA和Z-RNA结合,从而在病毒感染时促进坏死的激活91。
- 这些RHIM适配蛋白的激活还可以导致坏死效应蛋白MLKL的磷酸化、寡聚化和膜定位。
- ZBP1与RIPK3的RHIM依赖性结合被RIPK1的支架功能所抑制50,92。
- 因此,RIPK1和ZBP1可能竞争与RIPK3的结合。
- 敲除Zbp1或核心干扰素信号成分可以延长Ripk1-null小鼠的生存期,这表明在RIPK1缺失时,可能激活了依赖ZBP1的干扰素介导的反应。
Para_05
- 代谢应激和低氧也可以独立于TNF或TNFR185,93促进RIPK1和坏死性凋亡的激活。能量应激激活的AMPK作为一个代谢检查点94。
- 尽管激活的AMPK介导人RIPK1的S416和鼠RIPK1的S415抑制性磷酸化,但持续的葡萄糖剥夺可以促进RIPK1、RIPK3和MLKL的激活以诱导细胞死亡85。
- 此外,在常氧条件下,RIPK1的激活受EGLN蛋白质在多个脯氨酸残基,例如Pro195,的脯氨酰羟基化控制93。
- 这导致RIPK1激活被磷酸化的冯·希佩尔-林道病肿瘤抑制蛋白(pVHL)结合识别并抑制(一种已知针对转录因子低氧诱导因子1α的E3泛素连接酶,是细胞对氧浓度反应的关键调节因子)进行蛋白水解酶降解93,95。
- 长期低氧使EGLN蛋白质失活,并且RIPK1上的脯氨酸羟基化以及与pVHL1的结合丧失,这可以促进RIPK1的激活而不影响其降解。
- RIPK1与代谢和低氧的关键介质,如AMPK和pVHL的相互作用,可能有助于RIPK1在多个器官(包括大脑、心脏和肾脏)介导缺血损伤的潜在机制57,96,97,98。
Pyroptosis
Para_01
- 半胱氨酸天冬氨酸蛋白酶被发现介导一种称为焦亡的程序性坏死形式。
- 因此,焦亡代表了一种由专门遗传途径调节的哺乳动物细胞中的另一种程序性坏死细胞死亡类型。
- 半胱氨酸天冬氨酸蛋白酶1亚家族的激活,包括小鼠中的caspase-1和caspase-11以及人类中的相应caspase-1、caspase-4和caspase-5,促进了一种炎症反应,其中成熟的IL-1β是通过剪切前IL-1β以及其他下游半胱氨酸天冬氨酸蛋白酶如caspase-3产生的。
- 激活的caspase-1和caspase-11可以介导gasdermin D(GSDMD)的剪切,以去除GSDMD上的一个自抑制羧基末端域,使得N端片段能够在质膜上形成孔道,从而导致焦亡。
- 由16个GSDMD N端单体组成的孔道具有10-14纳米的内部直径,能够介导在活细胞中无需信号肽的细胞因子如成熟IL-1β的释放,以及坏死。
- 焦亡主要发生在专业的吞噬细胞中,包括巨噬细胞、单核细胞和树突状细胞,在受到细菌或病毒等病原体刺激,或由脂多糖和病毒DNA等病原体来源物质诱导时发生。
Fig. 3: Pyroptosis induction and mature pro-inflammatory cytokine (IL-1β) release.
- 细菌和病毒感染可促进小鼠体内caspase-1和caspase-11的激活,以及人体内caspase-1、caspase-4和caspase-5的激活,从而介导gasdermin D(GSDMD)的裂解,使其从抑制性的C端结构域(GSDMD-CT)释放出孔形成的N端结构域(GSDMD-NT)。
- 活化的caspase-8、caspase-3和caspase-7以及病原体蛋白酶也可以介导GSDMD的裂解。
- GSDMD-NT形成的孔可以介导活细胞中成熟IL-1β和IL-18的分泌,促进炎症,同时通过细胞溶解促进焦亡。
- LPS,脂多糖。
Para_02
- 通过gasdermin孔隙释放成熟的IL-1β等促炎因子,是经由基于电荷的机制进行的,这一过程独立于细胞死亡。
- 长期以来,介导无信号肽的细胞因子,例如成熟的IL-1β释放的机制一直是研究和争论的主题。
- 在人体死后病理样本中,观察到与淀粉样蛋白-β斑块密切相邻的、显然存活的微胶质细胞和星形胶质细胞呈现GSDMD裂解阳性染色,这表明GSDMD的N端域形成孔隙触发了炎症反应。
- 值得注意的是,在严重炎症的存在下,炎症反应和细胞死亡对于生物体的生存都是至关重要的。
- 例如,Casp11基因敲除小鼠对脂多糖诱导的致死性高度抵抗,这可能是由于抑制了GSDMD孔隙介导的成熟IL-1β和IL-18等促炎因子的释放,抑制了焦亡,或者两者都有。
- 未来需要基于生物标志物的研究,以更好地理解减低致死性的机制。
Para_03
- 气体dermin家族的其他成员,从GSDMA到GSDME以及pejvakin,这些基因编码在人类基因组中,也可以被细菌蛋白酶或哺乳动物的颗粒酶A切割。
- 由细菌SpeB介导的GSDMA切割在感染A群链球菌(GAS)的角质形成细胞中促进焦亡。
- 具有三种人类GSDMA同源基因三重敲除的突变小鼠对超毒力GAS菌株入侵变得敏感,这表明焦亡作为一种宿主防御机制的作用。
- 颗粒酶A介导的GSDMB激活是自然杀伤细胞杀菌活动的一部分,该活动被志贺菌的IpaH7.8靶标。
- 有趣的是,发现某些细菌编码具有孔形成活性的气体dermin同源物,用于防御噬菌体感染,这表明气体dermins可能是一种在进化上保守的古老宿主防御机制。
Dead cell removal by efferocytosis
Para_01
- 消除死亡细胞体对于防止细胞内物质的释放非常重要,这些物质可能作为损伤相关分子模式激活炎症反应,并可能导致自身免疫。
- 通过一种被称为吞噬作用的清洁机制,由专业的吞噬细胞如巨噬细胞和树突状细胞介导,有效地移除凋亡小体,对于避免这种自身免疫原性是非常重要的。
- 胱天蛋白酶介导的磷脂酰丝氨酸(PtdSer)在凋亡细胞表面的暴露,作为吞噬作用的识别基序。
- 在活细胞中,PtdSer通常由翻转酶,包括ATP11A和ATP11C维持在质膜的内叶。
- 由于胱天蛋白酶-3依赖性的切割和翻转酶的失活以及同时激活磷脂酰肌醇翻转酶XKR8,C. elegans中的CED-8同源物,PtdSer在凋亡细胞表面变得暴露。
- 巨噬细胞的吞噬作用涉及通过各种PtdSer受体如TIM4、TAM酪氨酸激酶受体、MFGE8、GAS6和蛋白质S对凋亡细胞的识别。
- 与C. elegans中观察到的情况类似,哺乳动物细胞中的Rho和Rab家族的小G蛋白及其鸟苷酸交换因子参与吞噬作用的执行,通过调节吞噬杯的形成和吞噬体的成熟以降解凋亡小体。
Para_02
- 与炎症小体激活的胱天蛋白酶一致,炎症细胞死亡时,其外质膜上也会展示磷脂酰丝氨酸‘吃我’信号,并且死细胞尸体迅速被吞噬细胞清除。
- 这些数据表明,凋亡细胞和炎症细胞都可以在体内被吞噬细胞消除。
- 由于坏死性凋亡不涉及胱天蛋白酶的激活,目前尚不清楚坏死性凋亡的细胞尸体如何在体内被清除。
- 理解程序性坏死,包括坏死性凋亡和炎症小体,的吞噬机制很重要,因为高效吞噬机制的存在可能会导致在细胞溶解和细胞内损伤相关分子模式(DAMPs)释放之前,吞噬并移除垂死细胞。
Membrane rupture after cell death
Para_01
- 坏死或无法有效清除凋亡细胞体可能导致质膜破裂,这一过程涉及膜蛋白NINJ1125。
- 在垂死细胞中,NINJ1蛋白会与紧密排列成栅栏状数组的跨膜α-螺旋126形成丝状结构。
- 在肝脏损伤的动物模型中应用抗-NINJ1抗体可以限制组织损伤,这表明针对膜损伤可能改善疾病的病理127。
- NINJ1可能通常参与功能性突触的维持,因为Ninj1-null小鼠表现出强迫性理毛引起的脱发、自我造成的损伤以及增加的焦虑样行为128。
Para_02
- NINJ1 已被证实能在周围神经系统和中枢神经系统(CNS)损伤后介导细胞间粘附。
- 当轴突受伤时,背根神经节神经元的 NINJ1 水平会升高,并且通过顺向运输到损伤部位。
- NINJ1 也可能通过促进免疫细胞的活化和免疫细胞穿越血脑屏障参与多发性硬化和中风模型中的炎症反应。
- 需要进一步的研究来了解 NINJ1 在调节这些状况中的作用是否涉及细胞膜的破坏和细胞死亡。
- NINJ1 介导的质膜破裂可能涉及急性神经组织和组织损伤后的病理过程,这些过程伴随着大量细胞死亡,可能会‘超过’死亡细胞清除机制的负荷。
- 另外,吞噬作用的遗传缺陷可能会降低死亡细胞清除的效率,导致破裂细胞膜的死亡细胞积累。
Interaction of programmed cell death pathways
Para_01
- 尽管凋亡、焦亡和坏死是由基因指定的细胞死亡机制执行的,但凋亡、焦亡和坏死在生物体中的进化起源和功能却大相径庭。
- 调节凋亡的基因存在于诸如线虫和苍蝇等原始生物中,而调节坏死如RIPK1、RIPK3和MLKL的基因仅在高度进化的脊椎动物和哺乳动物(如小鼠和人类)的亚群中发现。
- Apaf1缺陷小鼠、Casp9缺陷小鼠以及Casp3和Casp7双重缺陷小鼠的胚胎发育缺陷显示了凋亡在发育中的功能重要性。
- 相比之下,Casp1缺陷小鼠和Casp11缺陷小鼠是健康的,对炎症性凋亡和焦亡有抗性,并且不释放成熟的IL-1β和IL-18。
- 此外,虽然Ripk1缺陷小鼠会在围产期死亡,但多个表达催化活性失活的RIPK1激酶的突变小鼠品系却是正常的。
- Ripk3缺陷小鼠和Mlkl缺陷小鼠也是正常的。
- 进一步地,阻断RIPK1激酶和坏死可以抑制Casp8缺陷小鼠、Fadd缺陷小鼠、Abin1缺陷小鼠和Tbk1缺陷小鼠的早期胚胎致死性,这表明坏死可能参与清除发育不良的胚胎。
- 因此,凋亡参与调节发育,而坏死可能已经进化为通过淘汰高等脊椎动物中的缺陷胚胎来保护正常发育。
Para_02
- 在癌症中发现了凋亡和坏死的不同作用。FADD的扩增和表达增加与许多类型的癌症进展有关,包括乳腺癌、卵巢癌和肺癌。
- 相反,许多癌症和癌细胞系表现出RIPK3表达减少或丧失,与坏死抑制有关。
- 这些发现表明,凋亡支持生物体发育,而坏死作为一种检查点,在胚胎发育和成人生活中caspases调节异常时,阻止疾病状态的进展。
- 特别是,癌细胞中普遍存在的RIPK3表达丧失提示坏死在癌症发展中的作用作为一种检查点。
Autophagy and lysosomal cell death
Para_01
- 在以下部分,我们将讨论由于破坏了不涉及遗传编程的细胞死亡机制的稳态促生存机制而导致的细胞死亡的典型例子。
- 由于细胞内稳态失衡而死亡的细胞也可能表现出某些凋亡或坏死样特征。
- 然而,在这些条件下,仅抑制凋亡或坏死本身可能不足以拯救细胞生存,因为关键的细胞稳态促生存机制已经丧失。
Para_02
- ‘自噬性细胞死亡’这个术语最早被用来描述一种在胚胎组织中与凋亡形态学上不同的细胞死亡形式。"," Sentence_02 ":"自噬性细胞死亡已经在果蝇的变态过程中被描述。"," Sentence_03 ":"然而,自噬是一种正常的生理机制,它将细胞质内容物输送到溶酶体进行降解,并在压力下促进细胞存活。"," Sentence_04 ":"抑制自噬可以导致坏死。"," Sentence_05 ":"此外,自噬激活的形态学证据通常与细胞死亡过程(如坏死性凋亡)有关。"," Sentence_06 ":"阻断自噬对细胞死亡的影响最小,但会增加与坏死性凋亡相关的细胞碎片的积累,这表明在坏死性凋亡中激活自噬会促进细胞碎片的降解,而不是细胞死亡。"," Sentence_07 ":"在神经系统中,自噬对于通过促进聚集倾向蛋白质和功能障碍的细胞器(如损伤的线粒体)的降解来维持细胞内稳态至关重要。"," Sentence_08 ":"自噬或溶酶体功能的缺陷与与衰老相关的神经退行性疾病有关。
Para_03
- 溶酶体是一个重要的细胞内回收中心,充满了不同的水解酶,如溶酶体组织蛋白酶,这些酶能降解细胞大分子,产生氨基酸和其他代谢产物以供回收,因此,溶酶体对细胞生存至关重要。
- 然而,这个细胞器也被称为‘自杀囊’,因为溶酶体膜破裂的后果是致命的,由于溶酶体组织蛋白酶的泄漏,可能导致溶酶体细胞死亡。
- 溶酶体细胞死亡是通过组织蛋白酶介导的关键凋亡调节因子的裂解和激活来执行的,如半胱氨酸天冬氨酸蛋白酶家族和BCL-2家族成员,以及组织蛋白酶对关键生存机制的直接影响。
- 戈谢病是由GBA基因(编码葡萄糖脑苷脂酶(GBA))突变引起的最常见的溶酶体储存疾病。
- 在神经元中条件性丢失GBA的小鼠会导致RIPK1和RIPK3的表达增加和非凋亡细胞死亡,这种死亡可以通过Ripk3基因敲除来减弱。
- 此外,由脊髓损伤诱导的溶酶体损伤可能介导RIPK1招募到溶酶体,并促进RIPK1和RIPK3的激活。
- 由于脂褐素的积累导致的溶酶体渗透性增加,是导致Stargardt病和干性年龄相关性黄斑变性中视网膜色素上皮细胞退变的原因,可以通过激活MLKL导致非典型的坏死性凋亡。
- 此外,还有一大组溶酶体储存疾病,这些疾病是由这个细胞器功能改变引起的,并在多个器官,尤其是中枢神经系统中表现出严重的症状。
- 溶酶体稳态细胞生存机制的丧失可能是这些患者细胞死亡的主要驱动力。
Fig. 4: Cell death mediated by disruption of lysosomal function.
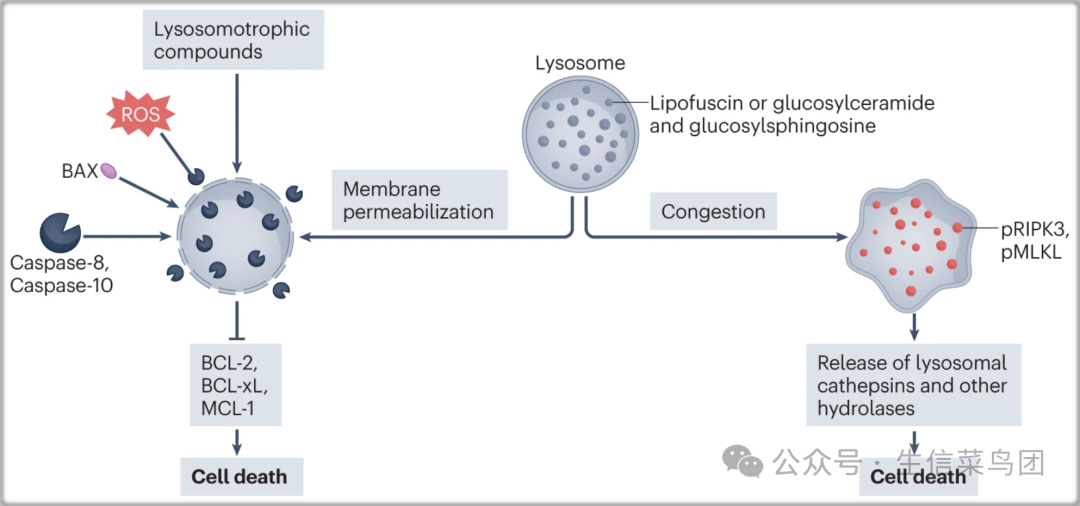
- 溶酶体膜透化(左),可能是由活性氧种(ROS)、溶酶体营养化合物以及活化的BAX和胱天蛋白酶诱导的,导致组织蛋白酶和其他水解酶从溶酶体腔释放到细胞质中。
- 溶酶体拥堵(右),是由Gaucher病中的葡萄糖神经酰胺和葡萄糖鞘氨醇积累以及Stargardt病和干性年龄相关性黄斑变性中的脂褐素积累诱导的,可以导致RIPK3和MLKL的激活。
- pMLKL,磷酸化的MLKL;pRIPK3,磷酸化的RIPK3。
Entosis
Para_01
- 胞吞现象发生时,一个细胞会吞没并消化另一个活细胞;因此,它可能被认为是一种细胞食人现象。胞吞过程的启动是由被吞没细胞与细胞外基质的脱离引起的,并且涉及由肌动蛋白、肌球蛋白II、Rho-GTP酶和Rho相关蛋白激酶 ROCK 产生的收缩活动。最终,内化的细胞通过自噬脂质化介导的溶酶体过程被降解,这一过程与内化病原体如单核细胞增生李斯特菌、A群链球菌和志贺氏菌的降解过程有相似之处。在葡萄糖饥饿条件下激活AMPK可以增加胞吞的频率,这表明代谢应激在调节胞吞过程中发挥作用。此外,TRAIL激活DR4和DR5也可以通过caspase-8的支架功能刺激胞吞,而不是通过其蛋白裂解活性。胞吞现象在人类癌症中存在,但胞吞在癌症中的作用需要进一步研究。
Ferroptosis
Para_01
- 铁死亡是由铁依赖的脂质过氧化介导的,最初被定义为可以通过小分子如erastin激活的细胞死亡,erastin是一种对胱氨酸/谷氨酸反向转运蛋白xCT的不可逆抑制剂,或者是RSL3,一种GPX4抑制剂。铁死亡在形态上不同于凋亡或坏死,并不涉及介导凋亡或坏死细胞机制的细胞器。
- 维持细胞稳态的氧化还原平衡对细胞和动物存活至关重要。胱氨酸/谷氨酸反向转运蛋白xCT,由SLC7A11基因编码,和GPX4是细胞氧化还原机制的两个关键调节因子,它们保护细胞免受脂质过氧化的伤害。
- xCT是一种异二聚体氨基酸反向转运蛋白,专门用于将谷氨酸输出以换取胱氨酸的输入。胱氨酸的还原生成半胱氨酸,这是在谷胱甘肽合成中的限速前体,保护细胞免受氧化应激。
- GPX4是一种含硒蛋白质,具有依赖谷胱甘肽的过氧化物酶活性,是细胞对抗脂质过氧化的主要防御机制。GPX4的失活会导致活性氧种(ROS)的过量产生,引发脂质过氧化,破坏质膜和线粒体膜,从而激活细胞死亡。
- 在小鼠中可诱导GPX4失活,导致脂质氧化引起的急性肾功能衰竭和动物死亡。多不饱和脂肪酸的脂质过氧化,是细胞ROS增加的一个后果,产生反应性醛,如4-羟基壬烯醛(4-HNE),它能够以细胞类型和浓度依赖的方式与蛋白质、脂质和DNA形成加成物。
Fig. 5: Lipid peroxidation and ferroptosis.
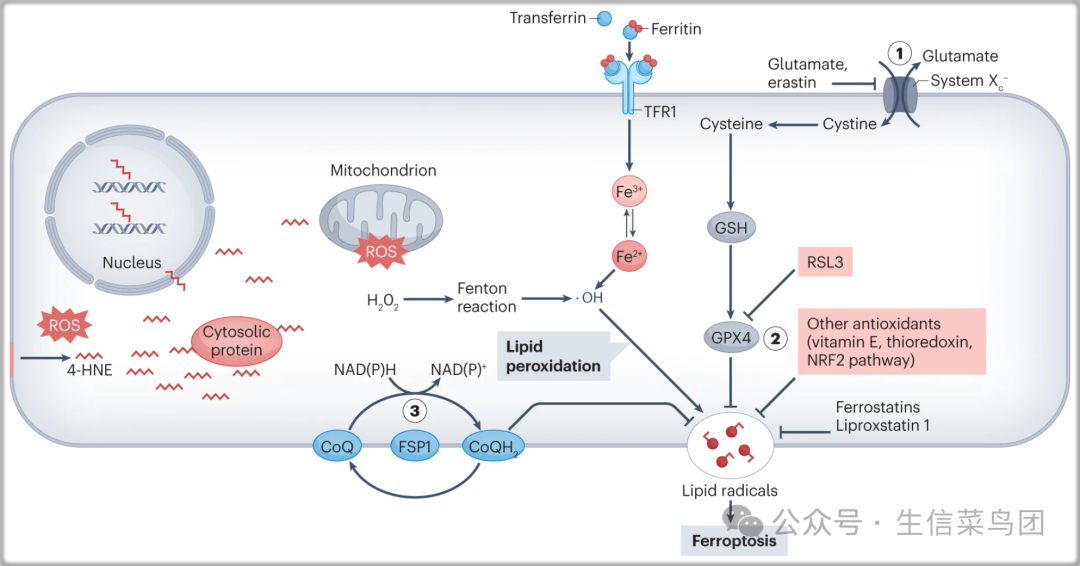
- 细胞机制保护脂质过氧化包括(1)xCT(由SLC7A11基因编码),它是一种异二聚体氨基酸反转运蛋白,专门用于以1:1的比例导入胱氨酸,胱氨酸的氧化形式,同时输出谷氨酸。
- 胱氨酸的还原生成半胱氨酸,是谷胱甘肽(GSH)合成的限速前体,保护细胞免受氧化应激。
- (2)GPX4,通过将脂质过氢化物转化为脂质醇来保护脂质过氧化。
- (3)定位在质膜上的FSP1(以前称为AIFM2),它作为一种氧化还原酶,将辅酶Q10(CoQ)还原生成亲脂性自由基捕获抗氧化剂,阻止脂质过氧化物。
- 多不饱和脂肪酸的脂质过氧化生成反应性醛,如4-羟基壬烯醛(4-HNE),可以与蛋白质、脂质和DNA形成相对稳定的加成物。
- 电子可能从细胞氧化还原反应中逃逸并被氧气捕获形成过氧化物(H2O2)。
- Fenton反应介导Fe2+和H2O2的氧化,产生羟基自由基与脂质反应形成脂质自由基,从而启动脂质过氧化。
- 铁死亡可以通过RSL3对GPX4的化学抑制或erastin对xCT的抑制来诱导,这会击败细胞对脂质过氧化的防御机制,通过大量铁依赖性脂质过氧化诱导细胞死亡。
- 铁稳定剂和脂氧稳定剂1通过作为自由基捕获抗氧化剂来抑制脂质过氧化。
- ROS,活性氧种;TFR1,转铁蛋白受体1。
Para_02
- 脂质过氧化可以作为细胞代谢的常态结果发生,当氧化剂攻击脂质,尤其是多不饱和脂肪酸时,这可以通过细胞氧化还原机制进行调节。
- 在炎症和疾病状态下,活细胞中的脂质过氧化增加。
- 随着年龄的增长,可能会导致氧化产物的产生增加,超出生物系统的解毒能力。
- 与年轻小鼠相比,老年小鼠的不同器官中通过HNEJ-1单克隆抗体检测到的4-HNE修饰蛋白水平升高,这表明衰老与脂质过氧化增加有关。
- 氧化应激和脂质过氧化长期以来与各种与衰老相关的人类疾病有关,包括糖尿病和神经退行性疾病。
- 然而,由于我们还没有针对铁死亡的特定生物标志物,因此应谨慎区分脂质过氧化(无细胞死亡)和铁死亡(有细胞死亡)。
Para_03
- 脂质过氧化水平受细胞氧化还原机制调节,作为一个可逆过程(图5)。通过增加NRF2介导的抗氧化防御基因表达,可以减少HNE修饰蛋白的积累。值得注意的是,弗里德里希共济失调,由编码铁蛋白的基因突变引起,会导致线粒体内铁处理改变和线粒体呼吸受损,这可以通过NRF2激活剂进行治疗。细胞自由基捕获抗氧化剂的产生,包括维生素E和维生素K,也可以提供对抗脂质过氧化和铁死亡的防御。除了GPX4,脂质过氧化还受到辅酶Q10氧化还原酶FSP1的调节,该酶催化再生具有自由基捕获抗氧化活性的泛醌还原形式(泛醇)。FSP1还可以将维生素K还原为氢醌,后者具有自由基捕获抗氧化活性。此外,性激素受体如雌激素受体和雄激素受体诱导的磷脂修饰酶MBOAT1和MBOAT2也可以调节脂质过氧化。
Para_04
- 过量的脂质过氧化产物积累可能会独立于细胞死亡而促进炎症。铁是细胞内稳态和生存的关键调节因子,因为它参与线粒体呼吸。
- 转铁蛋白受体1的丧失,该受体在大脑中铁的摄取中起作用,会导致神经元铁缺乏和多巴胺能神经元的进行性丧失。
- 铁蛋白轻链(FTL)基因的突变,该蛋白在铁储存中具有重要作用,会导致由于大脑铁过载而引起的罕见类帕金森病,称为神经铁蛋白病。
- 小胶质细胞中铁积累的炎症反应可能与铁死亡耦合。
- 因此,在抵御脂质过氧化的过程中,使关键的细胞还原-氧化 prosurvival 机制失活可能包括炎症的激活作为后果。
Excitotoxicity
Para_01
- 细胞内离子平衡的破坏,特别是特定离子通道过度激活后,可能导致一种被称为兴奋毒性的坏死类型(图6)。
- 在C. elegans中进行的关于机械敏感性的遗传研究,提供了一个优雅的原型例子,即由离子通道过度激活介导的细胞存活稳态机制的丧失所引起的细胞死亡187,188。
- mec-4和deg-1基因中的错义突变,这些基因编码的蛋白质与脊椎动物对氨力农敏感的上皮Na+通道的亚单位相似,导致了C. elegans中触觉感受器神经元的离子失衡、肿胀和坏死变性。
- 由突变MEC-4和DEG-1介导的细胞死亡独立于CED-3,但部分由天冬氨酸和钙蛋白酶介导,这些酶是由增加的细胞内Ca2+激活的(参考文献189,190)。
Fig. 6: Necrosis and excitotoxicity induced by disruption of homeostatic ionic balance.
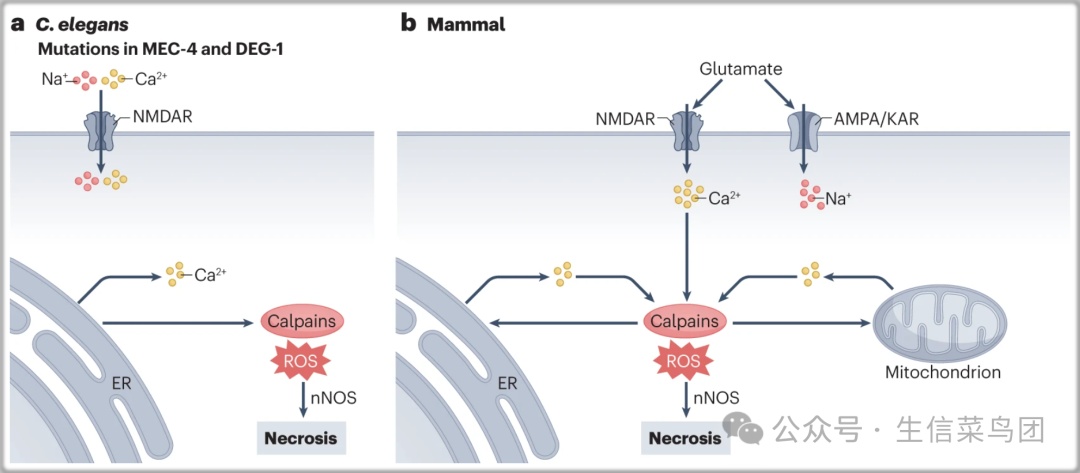
- MEC-4和DEG-1的过度激活会破坏细胞内离子平衡,这两个基因是与脊椎动物对阿米洛利敏感的上皮钠通道的同源基因,这会导致秀丽隐杆线虫的触觉感受神经元发生坏死性变性。
- 这种细胞死亡不依赖于CED-3,但部分由天冬氨酸和钙蛋白酶介导,这些酶是由细胞内Ca2+的增加所激活的。
- 在哺乳动物神经系统中,病理条件下谷氨酸水平的升高也可能导致细胞内Ca2+超载和细胞生存的丢失,这是由于离子失衡所致。
- 兴奋毒性的下游细胞死亡效应器可能包括凋亡和坏死性凋亡过程,并且可能取决于稳态破坏的程度。
- ER代表内质网;nNOS代表神经元一氧化氮合酶;ROS代表活性氧种。
Para_02
- 谷氨酸是哺乳动物神经系统中关键的兴奋性神经递质,能够驱使Ca2+流入神经元。在病理状态下,改变的Ca2+信号可能导致这种第二信使的水平过高,从而由于离子失衡引发兴奋性毒性。细胞在遭受细胞内Ca2+超负荷时无法存活。兴奋性毒性的下游细胞死亡效应器可能包括凋亡和坏死过程,并且可能取决于这种稳态破坏的程度。星形胶质细胞对谷氨酸的清除或回收也可能决定谷氨酸作为神经递质的可用性,以进行适当的信号传导和预防神经元的过度兴奋。过度的或持续的激活谷氨酸门控离子通道,如NMDA受体,已被涉及在急性神经元损伤(如脑缺血、癫痫持续状态、创伤性中枢神经系统损伤和低血糖)后中枢神经系统中的坏死细胞死亡。谷氨酸的细胞毒性可能被增加的Ca2+流入促进神经元一氧化氮合酶激活的能力所介导,而神经元一氧化氮合酶也已知调节许多生理功能,例如突触可塑性、学习、记忆和神经发生。神经元中的兴奋性毒性事件也能改变氧化状态,这已被证明可以驱动S-亚硝基化。
Mitotic catastrophe
Para_01
- 有丝分裂灾难是一种由化学或物理压力导致的非适当有丝分裂进入引起的细胞死亡类型。
- 从生物化学角度看,有丝分裂灾难的特点是未复制或受损的染色体在着丝粒处发生双链DNA断裂的过早凝聚。
- 具有G2检查点基因遗传缺陷的癌细胞,如ATM、ATR、CHEK1、CHEK2和 polo样激酶基因(PLK1、PLK2、PLK3),可以通过针对微管的治疗药物如紫杉烷、长春花生物碱和秋水仙碱以及DNA损伤剂诱导其发生有丝分裂灾难。
- 经历有丝分裂灾难的癌细胞可能会显示出caspase激活的特征。
- 然而,抑制caspase活性并不能阻止缺陷有丝分裂染色体的分离,这可能导致细胞分裂缺陷和染色体数目异常。
Cell death in human diseases
Para_01
- 在本节中,我们将简要讨论细胞死亡和细胞内稳态破坏在人类疾病中的作用,而框2则讨论了对程序性细胞死亡机制理解的提高对药物发现的影响。
[div_box]
Anti-apoptotic BCl-2 family as oncogenes
抗凋亡的BCl-2家族作为致癌基因
Para_01
- BCL-2在人类的滤泡性淋巴瘤中由于t(14;18)染色体重排而转录激活,并且许多类型的癌细胞中也发现了抗凋亡BCL-2家族成员的过表达。
- 抗凋亡BCL-2家族的促癌活性是通过在压力条件下延长癌细胞的存活时间,而不是促进细胞增殖。
- 这一家族在调节死亡与存活之间的平衡中具有关键作用,这一点得到了venetoclax(BCL-2特异性抑制剂)和navitoclax(BCL-2、BCL-xL和BCL-W的抑制剂)成功开发用于治疗白血病和淋巴瘤的证实。
Necroptosis in neurodegenerative diseases
神经退行性疾病中的坏死
Para_01
- 坏死性凋亡已被证实参与多种人类疾病的发生机制,这些疾病的特点是细胞死亡伴随炎症,如缺血性脑损伤、青光眼、多发性硬化症(MS)和肌萎缩性侧索硬化症(ALS)。
- 在ALS、MS和阿尔茨海默病患者死后病理样本中,已鉴定出RIPK1、RIPK3和MLKL的激活生物标志物,包括磷酸化形式(pRIPK1、pRIPK3和pMLKL)。
- 由于RIPK1仅通过DD介导的相互作用被招募到TNFR1,因此,RIPK1的激活特异性地介导TNFR1信号传导。
- 因此,抑制RIPK1为我们提供了一个机会,在不影响TNFR2的情况下,选择性地抑制由TNFR1介导的有害反应。
Inflammation mediated by RIPK1 and caspases
RIPK1和caspases介导的炎症
Para_01
- 两种主要的促炎途径包括RIPK1介导的TNFR1下游促炎细胞因子的产生以及炎症小体介导的caspase-1活化。
- 尽管这两种途径的上游激活因子可能显示出刺激物和细胞特异性的差异,但下游细胞因子的产生和释放通常包括一组共同的促炎因子,如TNF、IL-1β、IL-6和IFNγ,然而这种释放的水平、时机和组织特异性差异对于决定对生物体的影响可能是至关重要的。
- caspase-8介导的RIPK1切割的失活会导致人类自身炎症性疾病。
- 在坏死性和炎症性条件下,如ALS中,已经检测到激活的RIPK1存在于细胞核中。
- 核RIPK1与一系列转录激活因子和协同激活因子相互作用,例如p65(也称为RELA)、SP1和JUNB,以及几乎所有BAF染色质重塑复合体的组成部分,通过调节染色质动力学来促进促炎因子的转录诱导。
Para_02
- caspase-1、caspase-11和相关caspases的激活是由多分子复合物介导的,这些复合物被称为炎症小体,包括不同的PRRs,如NLRP3、NLRP1、NAIPs、NLRC4、AIM2、Pyrin和诸如ASC之类的适配蛋白。
- 这些复合物的下游信号是由PRR依赖性和刺激依赖性方式决定的。
- 最近的研究发现,NLRP3炎症小体在神经退行性疾病的背景下被激活,导致炎症和变性。
- GSDMD的裂解调节成熟IL-1β和IL-18的释放,以促进炎症,这是人类炎症条件下的关键促炎机制。
Disruption of homeostasis in diseases
疾病中稳态的破坏
Para_01
- 蛋白质稳态的破坏,即蛋白质合成与降解之间的适当平衡,会导致神经退行性疾病中错误折叠和聚集蛋白的积累。
- 恢复细胞稳态是一个需要考虑的重要机制,特别是对于神经退行性疾病而言。
- 针对TRADD,TNFR1信号通路中的一个重要适配器,提供了一个机会来抑制依赖于RIPK1的凋亡,并激活自噬,通过促进错误折叠蛋白的降解来恢复细胞稳态。
Para_02
- 破坏氧化还原平衡会导致活性氧(ROS)的积累,这在慢性炎症疾病患者中是常见的。
- 此外,正如4-HNE水平的升高所指示的,脂质过氧化的积累可能导致细胞损伤和铁死亡。
- 由于大脑消耗的氧气量与体重比例失调(大约消耗全身20%的氧气,而大脑只占体重的约2%),因此大脑对氧化损伤极为敏感。
- 在患有遗忘型轻度认知障碍的患者大脑以及早期阿尔茨海默病的大脑中,脂质过氧化是一个早期的病理事件。
- 氧化应激和脂质过氧化可能涉及到促进阿尔茨海默病动物模型中淀粉样-β斑块的形成。
Conclusions and perspectives
Para_01
- 对线虫C. elegans的程序性细胞死亡进行的遗传学研究,首次揭示了控制发育性凋亡细胞死亡的分子机制(由EGL-1、CED-9、CED-3和CED-4介导)。
- 这种在进化上保守的细胞死亡机制在哺乳动物细胞中得到了显著扩展,可能是通过基因复制,包括了编码CED-3(半胱氨酸天冬氨酸蛋白酶家族)和CED-9(BCL-2家族)的多个同源基因,这些基因不仅介导发育性细胞死亡,还介导与疾病相关的细胞死亡和炎症。
- 由RIPK1、RIPK3、MLKL和ZBP1介导的坏死性凋亡的激活,可能是由于在复杂的多细胞生物中消除细胞以及在抵御病原体时产生的额外需求,而在进化后期才被加入。
- 此外,caspases(包括caspase-11、caspase-1、caspase-3和caspase-8)对GSDMDs的切割可以促进焦亡的发生,焦亡是一种坏死形式,它也可以在病理条件下介导无信号肽的细胞因子,如成熟的IL-1β的释放,从而驱动炎症。
Para_02
- 激活特定的细胞死亡和炎症途径在体内的后果可能因细胞类型和这些途径的不同介质表达水平而有所不同。
- 例如,RIPK1的激活与微胶质细胞中促炎细胞因子的产生增加有关,而不是细胞死亡,而RIPK1在寡突细胞中的激活可能导致细胞死亡和脱髓鞘,这是由于成熟寡突细胞的丧失,并导致新生寡突细胞的补偿性再生。
- 此外,caspase-1和caspase-11的激活可以促进下游caspases(caspase-3和caspase-7)的激活,因为它们的特异性与其他长前域caspases(如caspase-9)相似。
- 观察到caspase-1和caspase-11以及caspase-8和caspase-3可以裂解凋亡底物和焦亡底物,这与它们的裂解特异性一致。
- 然而,caspases的激活是否导致凋亡或坏死可能取决于在死亡细胞中被裂解的数百个关键caspase底物中的表达水平。
Para_03
- 多种细胞稳态机制参与介导细胞存活,这解释了发现的细胞死亡方式的复杂性和不断增加的数量。
- 由于细胞稳态的促存活机制的失活可能为选择性地诱导癌细胞死亡提供多种选择,因此,通过化学或基因操作靶向选择性的癌细胞偏爱的促存活机制,在未来可能为癌症治疗提供令人兴奋的新选择。
- 此外,尽管许多治疗干预策略都集中在阻断促死亡途径的异常激活上,但增强特定促存活机制以治疗神经退行性疾病的可能性应该进一步探索。
- 确定哪些细胞存活信号被中断,并开发特定的治疗策略来恢复它们,可能使得细胞例如神经元或胰腺β细胞,在特定疾病条件下的有害环境中存活下来。
- 因此,理解连接细胞稳态和细胞死亡的信号通路可能使得能够开发出新的治疗途径,这些途径能够抑制细胞死亡并恢复细胞稳态和存活。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-12-26,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号