不要简单的相信作者提供的表达量矩阵


GEO(Gene Expression Omnibus)数据库是一个公共的基因表达量数据库,它收录了来自不同平台的高通量基因表达数据,包括Affymetrix、Illumina和Agilent等。每个平台都有自己的文件格式和数据处理流程,以下是对这三个主要平台的简要介绍:
- Affymetrix:
- 平台特点:Affymetrix平台使用微阵列技术,每个探针对应一个特定的基因或转录本。
- 文件格式:Affymetrix数据通常以
.CEL文件格式存储,这是一种二进制格式,包含了原始的荧光强度值。 - 数据处理:需要使用专门的软件(如Affymetrix Power Tools, dChip, or R/Bioconductor的
affy包)来读取.CEL文件,并进行标准化和背景校正。
- Illumina:
- 平台特点:Illumina平台使用测序技术,可以提供单核苷酸多态性(SNP)和基因表达数据。
- 文件格式:Illumina数据以
.idat文件格式存储,这是原始的图像强度数据。 - 数据处理:需要使用Illumina自己的软件(如GenomeStudio)或其他第三方工具(如R/Bioconductor的
illuminaio包)来处理.idat文件,提取表达量数据,并进行标准化。
- Agilent:
- 平台特点:Agilent平台也使用微阵列技术,特点是可以灵活设计探针,适用于研究者自定义的基因集。
- 文件格式:Agilent数据通常以
.txt或.csv格式存储,包含了原始的荧光强度值。 - 数据处理:可以使用Agilent自己的软件(如Feature Extraction Software)或R/Bioconductor的
limma包等工具来处理这些文件。
处理这些平台的数据时,研究者需要了解各自平台的特点和数据处理流程,选择合适的工具和方法来进行分析。此外,由于不同平台之间的技术差异,直接比较不同平台的数据时需要格外小心,可能需要进行平台间的标准化或使用兼容的分析方法。
但是大部分情况下,我们偷懒会直接下载GEO数据库里面的作者上传的表达量矩阵,我们拿GSE13904举例说明,简单的代码如下所示:
library(AnnoProbe)
library(GEOquery)
getOption('timeout')
options(timeout=10000)
#获取并且检查表达量矩阵
##~~~gse编号需修改~~~
gse_number <- 'GSE13904'
dir.create(gse_number)
setwd( gse_number )
getwd()
list.files()
if(T){gset <- geoChina(gse_number)}
#gset = getGEO("GSE13904", destdir = '.', getGPL = F)
gset[[1]]
其实上面的代码就是远程读取:https://ftp.ncbi.nlm.nih.gov/geo/series/GSE13nnn/GSE13904/matrix/ 里面的文件:
- https://ftp.ncbi.nlm.nih.gov/geo/series/GSE13nnn/GSE13904/matrix/GSE13904_series_matrix.txt.gz (2024-10-08 05:42 51M)
这个文件是作者上传的表达量矩阵,所以数据预处理取决于作者的想法!
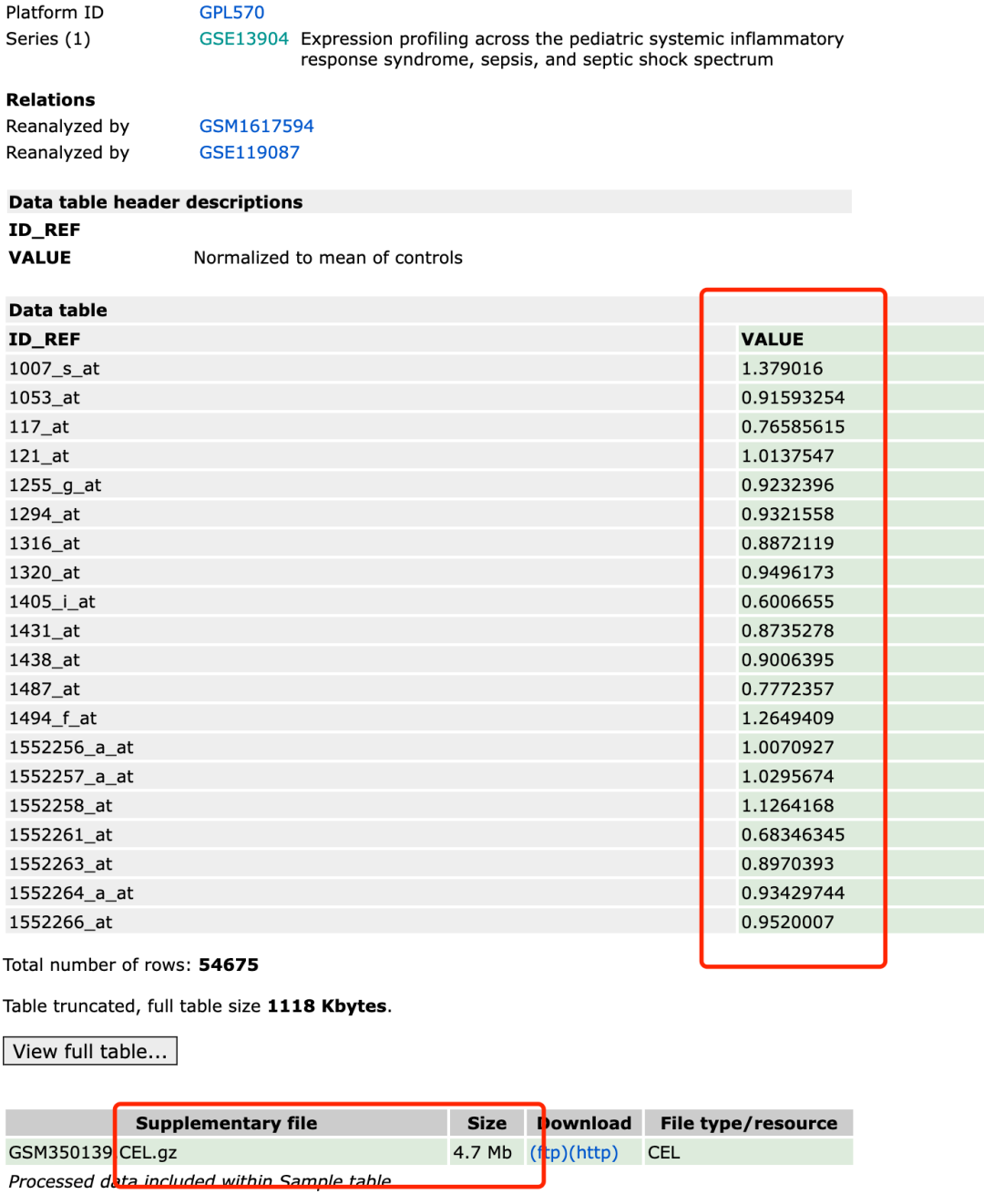
有一些时候会出现一些奇怪的矩阵,比如这个GSE13904数据集 ,可以看到 :
> a=gset[[1]]
> dat=exprs(a) #a现在是一个对象,取a这个对象通过看说明书知道要用exprs这个函数
> dim(dat)#看一下dat这个矩阵的维度
[1] 54675 227
> dat[1:4,1:4] #查看dat这个矩阵的1至4行和1至4列,逗号前为行,逗号后为列
GSM350139 GSM350140 GSM350141 GSM350142
1007_s_at 1.3790160 1.0096979 1.0634007 1.0500925
1053_at 0.9159325 0.6084510 1.2112615 0.8101592
117_at 0.7658561 0.7135977 1.0033835 1.1766533
121_at 1.0137547 1.5091416 0.9252635 1.2368684
> range(dat)
[1] 0.0100 990.7652
# 每个样品的四分位数如下所示:
> apply(dat[,1:4], 2,fivenum)
GSM350139 GSM350140 GSM350141 GSM350142
[1,] 0.02240257 0.02496001 0.1536448 0.04565957
[2,] 0.87158622 0.76531896 0.8887694 0.87434662
[3,] 0.97803104 0.98077905 1.0041255 0.99884206
[4,] 1.08294430 1.16814570 1.1577705 1.12798295
[5,] 16.43987000 10.30501500 25.7737870 9.34895900
有点意思啊, 绝大部分样品都是中位值居然都是1附近,这个就不符合我们的认知。正常情况下,我们的表达量芯片得出来的矩阵里面的数字范围应该是0到15直接,大部分在5到8附近。
如果我们直接从这个GSE13904数据集里面的找到脓毒症和正常对照,这两个分组的样品,然后试试看做差异分析 :
pd=pData(a)
kp1=grepl('Sepsis',pd$title);table(kp1)
kp2=grepl('Control',pd$title);table(kp2)
pd=pd[kp1 | kp2,]
dat=dat[,kp1 | kp2]
library(stringr)
##~~~分组信息编号需修改~~~
group_list=ifelse(grepl('Control',pd$title ,ignore.case = T ),
"control","case")
table(group_list)
group_list <- factor(group_list, levels = c("control","case"))
table(group_list)
会出现很诡异的 差异分析结果 :
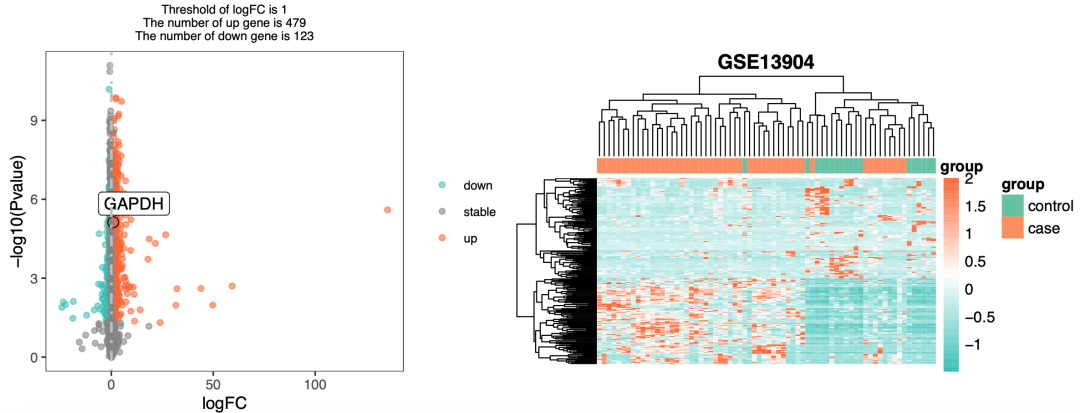
诡异的 差异分析结果
如果我们打开具体的一个样品,是可以看到它的处理方法:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM350139
- Commercial Affymetrix Human Genome U133 Plus 2.0 GeneChip (Affymetrix, Santa Clara, CA).
- GeneChip CEL files were subjected to RMA normalization using the GeneSpring GX 7.1.
本来呢,使用affymetrix公司的官方软件GeneSpring是很标准的操作,但是值得注意的是作者画蛇添足多了一个处理:
- The Raw CEL files were processed using the RMA (Robust Multichip Average) built in GeneSpring software.
- All the samples were then normalized to the median of the controls.
使得每个样品的每个基因的表达量不再具有跨数据集的可比性:
如果要做差异分析或者后续高级分析
都需要读取这个数据集提供的cel文件,做出来自己的表达量矩阵,示例代码是:
library(oligo)
library(ggplot2)
# 把cel文件存放在 ../cel_files/ 文件夹里面
celFiles <- list.celfiles('../cel_files/',listGzipped = T,
full.name=TRUE)
celFiles
options(BioC_mirror="https://mirrors.westlake.edu.cn/bioconductor")
exon_data <- oligo::read.celfiles( celFiles )
dim(exprs(exon_data))
exprs(exon_data)[1:4,1:4]
### 标准化(一步完成背景校正、分位数校正标准化和RMA (robust multichip average) 表达整合)
exon_data_rma <- oligo::rma(exon_data )
exp_probe <- Biobase::exprs(exon_data_rma)
exp_probe[1:4,1:4]
dat=exp_probe
dim(dat)#看一下dat这个矩阵的维度
dat[1:4,1:4] #查看dat这个矩阵的1至4行和1至4列,逗号前为行,逗号后为列
pd = pData(a)
head(pd)
学徒作业
完成这个GSE13904数据集的脓毒症和正常对照,这两个分组的样品,的差异分析,基于作者矩阵,以及基于cel文件的矩阵,做两次差异分析后对比一下结果。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-11-14,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号